
元素Sr 对铸态Mg-2Zn-1Mn 合金力学性能的影响
2023-06-17 07:18项戈丰
中国科技纵横 2023年7期
项戈丰
(北京科技大学,北京 100083)
可降解生物医用镁合金在生物金属材料中因其优异的力学性能、良好的生物相容性和可降解性而受到广泛关注。然而,镁的快速降解速度仍然是其大规模应用的主要障碍。因此,迫切需要开发降解速率可控的可生物降解镁合金。
Mg-Zn-Mn 基合金,具有良好的生物相容性。该合金强度高、塑性好、加工硬化能力强,在生物结构材料特别是血管支架方面具有很大的应用潜力。但Mg-2Zn-1Mn 合金其综合力学性能以及耐蚀性与国际先进的生物医用镁合金相比还是略逊一筹。因此本文致力于在Mg-2Zn-1Mn 基合金的基础上,加入适当的Sr 元素,以改善其性能。
1.实验材料及方法
1.1 材料
本实验熔炼的镁合金所采用的原料有高纯镁(99.99wt.%),高纯锌粒(99.995wt.%),Mg-15wt.%Mn 中间合金,Mg-20wt%Sr 中间合金。实验设备采用新型SJ2 井式电阻炉,熔炼过程中采用CO2+SF6(99:1)混合气体作为保护气,在电阻炉中制备了名义成分为Mg-2.0Zn-1.0Mn-xSr(x=0、0.2、0.4、0.6)的合金铸锭。熔炼过程中首先将镁锭熔化,然后依次加入中间合金,760℃保温30min,然后降温至720℃浇注到石墨铸型中,结束熔炼工艺,浇注得到铸锭大小为Φ54×150mm。熔炼完用电感耦合等离子体发射光谱仪进行成分分析,所得结果如表1 所示。
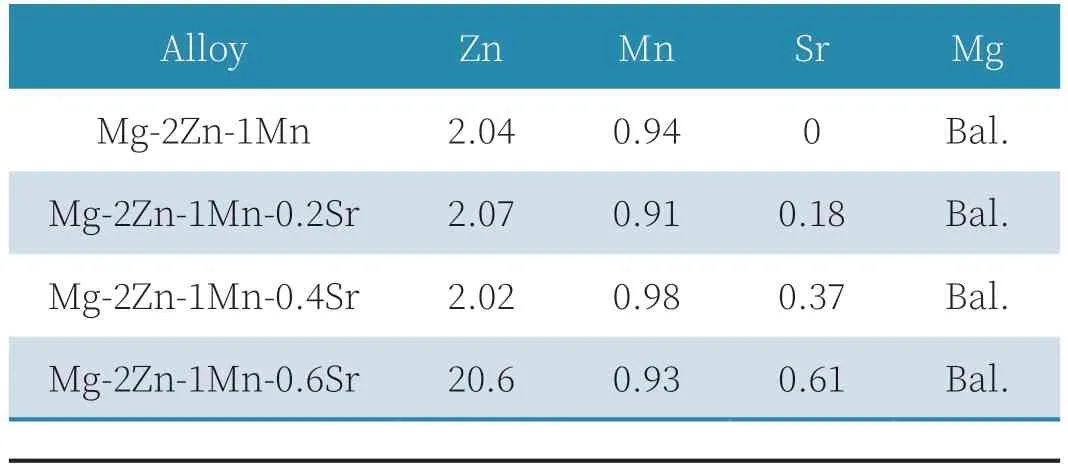
表1 合金的实际成分(wt.%)
1.2 微观组织表征
在铸锭中心部分取10mm×10mm×5mm 的块状试样,然后用800#、1200#、1500#、2000#、3000#、5000#的水磨砂纸按照从粗到细依次进行磨样,每次更换砂纸需要旋转90°并保证上一道次的划痕磨掉,然后在抛光机上进行酒精抛,待表面光亮成镜面状即可。进行金相观察前先对试样用20%的硝酸酒精化学抛光,然后用4%硝酸酒精浸蚀,时间为10s,采用光学显微镜、扫描电镜观察合金的显微组织,利用能谱仪分析合金微区的化学成分,利用X 射线衍射仪(XRD)对试样进行物相分析。
1.3 机械性能
根据GB/T 16865-2013 制取镁合金的室温拉伸试样,制成标距为25mm,宽度为6mm,厚度为2mm 的板材拉伸试样,采用拉伸试验机测试合金的室温力学性能,并用扫描电镜观察合金的拉伸断口形貌。
2.结果与讨论
2.1 Sr 对铸态Mg-2Zn-1Mn 合金组织的影响
图1 为Mg-2Zn-1Mn-xSr 合金的铸态组织图,(a)、(b)、(c)和(d)分别表示添加0wt.%、0.2wt.%、0.4wt.%、0.6wt.%不同Sr 含量的合金显微组织金相图。合金a 平均晶粒尺寸为101.12μm,晶间第二相较少,仅有些点状组织弥散分布在晶界上。随着Sr 元素的加入晶间第二相逐渐增多,分布更加均匀,晶粒尺寸明显变小,平均晶粒尺寸达到76.92μm。当添加0.4wt.%Sr 时,平均晶粒尺寸变为108.56μm,沿晶内或晶界分布的第二相数量明显增多。随着Sr 量的进一步增加,当添加0.6wt.%Sr 时,晶粒尺寸变化不明显,平均晶粒尺寸为107.92μm,晶间第二相组织粗化。随着Sr 含量的增加,由于一个α-Mg晶粒内形成若干枝晶,第二相沿枝晶边界强烈析出,因此第二相数量和面积都发生显著的变化。
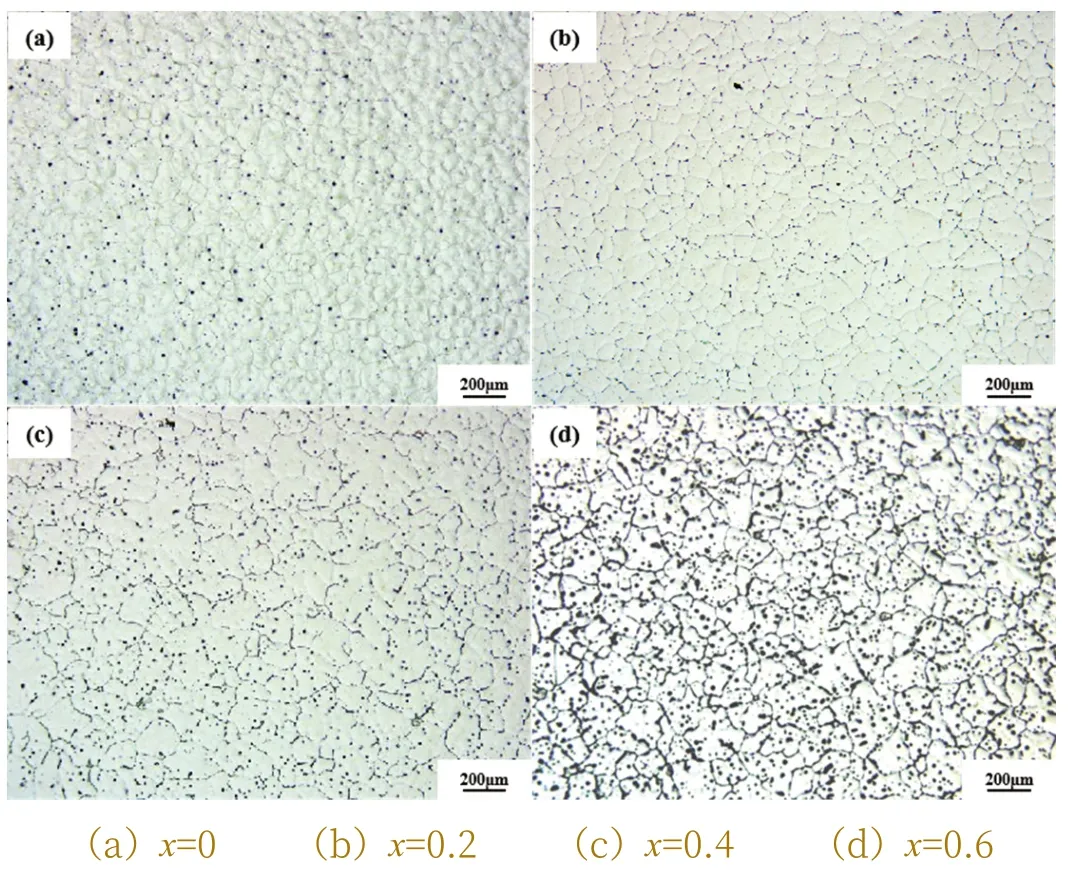
图1 铸态Mg-2Zn-1Mn-xSr合金金相组织
Sr 元素的加入对微观组织的影响主要因为其在Mg 中的固溶度较低(约等于0.11wt.%)以及非平衡凝固的并行效应[1]。一方面,在铸造过程(非平衡凝固)中,溶质中的Sr 原子被推向固/液界面前沿,导致固液界面前沿由于溶质再分配产生成分过冷,使得溶质的扩散发生缓慢,从而限制了晶体的生长速率,产生生长限制效应[2];另一方面,富锶液体沿着晶界积累,在那里它通过共晶反应固化,形成第二相组织,第二相组织对晶粒的长大具有钉扎阻碍作用。而且Sr 具有较大的生长抑制因子(GRF=3.51),因此在凝固过程中会形成许多枝晶,枝晶在生长过程中会产生更多的分枝,分枝逐渐增多并依次相连形成二次枝晶,从而将未凝固的液相分隔成许多细小而封闭的孤岛,导致溶质原子扩散速率减慢,从而导致晶粒生长速率减小,使得晶粒细化。并且由于生长抑制效应使得大量形核剂在固液界面前沿被激活,形核数量增多,从而使晶粒进一步细化。
图2 是铸态Mg-2Zn-1Mn-xSr 合金的X 射线衍射图谱。根据图谱分析,当未添加Sr 元素时,合金XRD 图谱只显示出了一定数量的镁峰。加入Sr 元素后,除了原本的镁峰外,还出现了Mg17Sr2相的峰值。因此判断Mg-2Zn-1Mn-xSr 合金主要由α-Mg 相和Mg17Sr2相构成,晶间和晶内析出的第二相被XRD 图谱确认为Mg17Sr2相,尤其是在Sr 含量最高的合金ZM21-0.6Sr 中尤为显著。综上分析,加入合金中的Sr 元素,极少地溶于镁基底形成固溶体,大部分Sr 和Mg 结合形成化合物Mg17Sr2相。由于Zn、Mn 含量较少,远低于其在镁中的固溶度,所以全部融入镁中形成固溶体,所以其余的峰值均显示为α-Mg 峰。另外,较大的尺寸会削弱XRD 的统计性,从而使α-Mg的一些峰消失[3]。

图2 铸态Mg-2Zn-1Mn-xSr合金X射线衍射分析
铸态Mg-2Zn-1Mn-xSr 合金的扫描电子显微镜成像图片如图3 所示,合金a 主要由α-Mg 和少量析出在晶内和晶界处的孤立的颗粒(直径约为1μm)第二相构成。加入元素Sr 后,一些第二相在晶粒内部(颗粒形状)和沿枝晶边界(条状)分散沉淀,条状第二相含量较少且沿晶界分布均匀,晶粒内部的粒装第二相数量增多。随着Sr 含量的进一步增加,在晶界处析出更多的条状第二相,且各个板条相互连接形成连续或半连续的网状组织,同时晶粒内部颗粒状第二相数量显著增加。当Sr 含量达到0.6wt.%,第二相体积呈线性增加,晶界处形成连续且粗大的网状组织,晶粒内部也形成较大的颗粒状组织。第二相的存在形式逐渐从孤立粒子演变为不连续网格,然后演变为半连续网格。根据微观组织变化结合XRD 图谱分析,加入Sr 元素后,促进了化合物Mg17Sr2相的形成。

图3 铸态Mg-2Zn-1Mn-xSr合金扫描电镜组织
2.2 Sr 对铸态Mg-2Zn-1Mn 合金力学性能的影响
铸态Mg-2Zn-1Mn-xSr 合金力学拉伸性能如图4 所示。随着Sr 含量的增多,合金拉伸性能总体呈现先上升后下降的趋势。当未添加Sr 元素时合金抗拉强度为150.5MPa,屈服强度为41.7MPa,伸长率为11.4%。添加0.2wt.%Sr后,合金抗拉强度和伸长率显著增加,都分别达到了最大值201.6MPa 和13.8%,屈服强度略有提升也到达最大值50.2MPa,与未加入Sr 时的合金相比,抗拉强度提升了34%,伸长率提升了21%,屈服强度提升20.4%。随着Sr元素的增加,合金力学性能开始下降,其中抗拉强度先快速下降,当Sr 含量为0.4wt.%~0.6wt.%时,抗拉强度下降缓慢逐渐到达最低值145.8MPa,低于未加入Sr 时的合金强度,屈服强度下降缓慢,当Sr 含量为0.6wt.%达到最低值35.2MPa。伸长率随着Sr 元素的增加呈线性下降,当Sr 含量为0.6wt.%达到最低值10.7%。综上所述,当Sr 含量为0.2wt.%时,合金综合力学性能最优,当Sr含量为0.6wt.%时,合金综合力学性能最差。表2 为铸态Mg-2Zn-1Mn-xSr 合金拉伸性能数据。

图4 铸态Mg-2Zn-1Mn-xSr合金拉伸性能
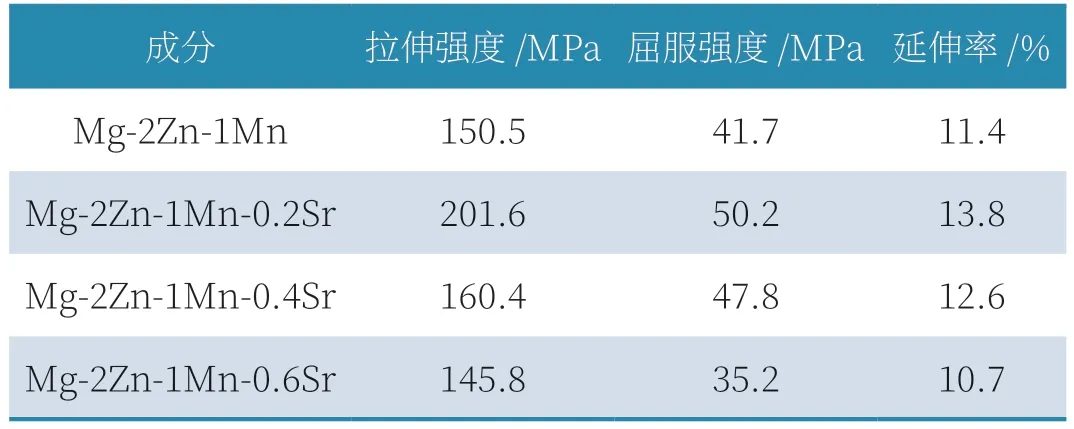
表2 铸态Mg-2Zn-1Mn-xSr合金拉伸性能
合金拉伸性能显著提升,这主要归因于加入微量元素Sr 后第二相的形成以及伴随的细晶粒和弥散强化效应。一方面引入Sr 元素后,所带来的生长限制效应,使得晶粒尺寸明显减小,产生细晶强化效应;另一方面在晶内所形成弥散的第二相组织,强化了镁合金基底,从而使合金综合力学性能显著提升。但加入过量Sr 后,由于在晶界处析出大量连续网格相Mg17Sr2,使合金脆性增加,综合力学性能降低。
3.结论
(1)随着Sr 元素的加入,合金平均晶粒尺寸明显变小,晶粒尺寸从101.12μm,减小到76.92μm,晶间第二相逐渐增多,随着Sr 元素的加入,析出在晶粒内部(颗粒形状)和沿枝晶边界(条状)的第二相为Mg17Sr2相。
(2)铸态Mg-2Zn-1Mn-xSr 合金力学拉伸性能随着Sr 含量的增多,总体呈现先上升后下降的趋势。Sr 含量为0.2wt.%时,合金综合力学性能最优。这主要归因于引入Sr 元素后,使得晶粒尺寸明显减小,产生细晶强化效应,其次在晶内所形成弥散的第二相组织,产生弥散强化效应,从而使合金综合力学性能显著提升。但加入过量Sr 后,由于在晶界处析出大量连续或半连续网格相Mg17Sr2,使合金脆性增加,力学性能下降。
猜你喜欢
汽车科技(2020年3期)2020-06-08
科学中国人(2017年35期)2017-06-08
电镀与环保(2016年2期)2017-01-20
材料科学与工程学报(2016年2期)2017-01-15
当代化工研究(2016年6期)2016-03-20
大型铸锻件(2015年1期)2016-01-12
中国质量与标准导报(2015年2期)2015-02-28
中国有色金属学报(2014年2期)2014-06-04
燕山大学学报(2014年2期)2014-03-11
中国有色金属学报(2012年10期)2012-09-26
