
新生儿钠牛磺胆酸共转运多肽缺陷病2例报道及文献回顾
2023-06-15 07:49:06丁海燕张连红
检验医学与临床 2023年11期
丁海燕,陈 琪,张连红△
1.武汉科技大学医学院,湖北武汉 430000;2湖北省天门市第一人民医院新生儿科,湖北天门 431700
钠牛磺胆酸共转运多肽(NTCP)缺陷病是由于溶质转运蛋白家族10成员1(SLC10A1)基因突变引起的一种遗传性胆汁酸代谢病,为一种常染色体隐性遗传病,NTCP 缺陷病患者体内血浆中结合胆汁盐进入肝细胞的主要转运体的功能受到影响[1-2]。主要临床表现为高胆汁酸、皮肤瘙痒和脂肪吸收不良,导致全面生长迟缓和脂溶性维生素缺乏等。目前,国内外相关文献报道并不多见,长期预后及临床表现不明确。湖北省天门市第一人民医院收治并确诊为NTCP缺陷病患儿2例,现将其临床资料报道如下,以提高临床医生对该病的认识。
1 临床资料
1.1病例1 患儿,男,出生后1个半月,因发现皮肤黄染40+d到湖北省天门市第一人民医院就诊。患儿出生后第2天出现皮肤黄染,在新生儿科住院治疗,期间予以蓝光光疗退黄,好转出院。出院后患儿黄疸反复出现,间断在门诊予以蓝光治疗,多次肝功能检查提示胆汁酸呈持续性升高,丙氨酸氨基转移酶(ALT)正常。见表1。患儿患病期间精神食欲可,大、小便正常,生长、发育正常。患儿系第 2 胎第 1 产,自然受孕,胎龄33+3周在产科顺产出生,出生体重2.35 kg,头围31 cm,身长43 cm,均位于同孕龄同性别儿胎儿50%左右,生后无窒息史,否认家族遗传病史及传染疾病史。生后因“早产”在新生儿科住院治疗,出生后1个月时好转出院。就诊时查体:体重4 kg,颜面部及躯干部皮肤轻度黄染,巩膜未见黄染,前囟平软,心脏、肺部、腹部、神经系统查体均未见异常,四肢活动可。辅助检查:肝、胆、胰、脾彩色多普勒超声检查未见异常,甲状腺功能检查未见异常,TORCH检查提示巨细胞病毒免疫球蛋白G阳性,心脏、头颅彩色多普勒超声检查均未见异常。因患儿肝功能检查提示胆汁酸呈持续升高状态,与其家属沟通后于2022年3月26日完善遗传性黄疸单病包基因检测。2022年4月11号回报:SLC10A1(NM 003049.4):c.800 C>T (p.S267F),纯合突变,患儿母亲为该变异的杂合携带者,患儿父亲该位点未检出。后期陆续完善脂溶性维生素及微量元素检测,结果提示锌及维生素A缺乏。患儿于1岁时复查胆汁酸恢复正常。
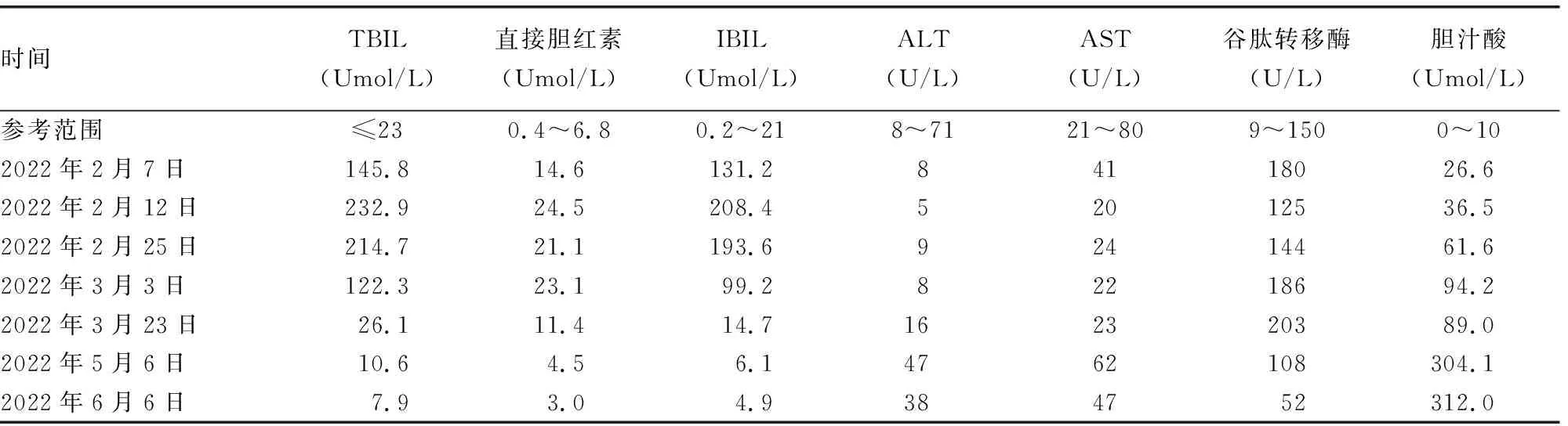
表1 病例1患儿肝功能结果
1.2病例2 患儿,女,出生后1个月,因胆汁酸异常到湖北省天门市第一人民医院就诊进行肝功能复查,患儿出生后因羊水Ⅲ度污染在新生儿科住院治疗,因住院期间肝功能检查提示胆汁酸偏高,故嘱患儿出院后复查,患儿出生后1个月时复查肝功能提示胆汁酸呈持续性升高,TBIL、IBIL、ALT、AST均正常。见表2。患儿未出现特殊不适。患儿系第 1 胎第 1 产,为自然受孕,胎龄39+6周在产科顺产娩出,出生体重3.65 kg;否认家族遗传疾病史,否认传染病史,父母非近亲婚姻。生产时羊水呈Ⅲ度粪染,出生后患儿因呻吟、吐沫伴呼吸困难10 min收入新生儿科治疗。患儿入院后予以呼吸支持、抗感染及蓝光治疗等对症治疗,住院9 d好转出院。患儿出院后定期在门诊复查肝功能提示胆汁酸呈持续升高状态,与其家属沟通后于2022年6月24日完善遗传性黄疸单病包基因检测。2022年7月15号回报:SLC10A1(NM 003049.4):c.800 C>T (p.S267F),杂合突变,患儿父亲为该变异的杂合携带者;SLC10A1(NM 003049.4):c.263T>C (p.I88T),杂合突变,患儿母亲为该变异的杂合携带者。本例患儿生长、发育正常,后期陆续完善脂溶性维生素及微量元素检测,结果均未见异常。
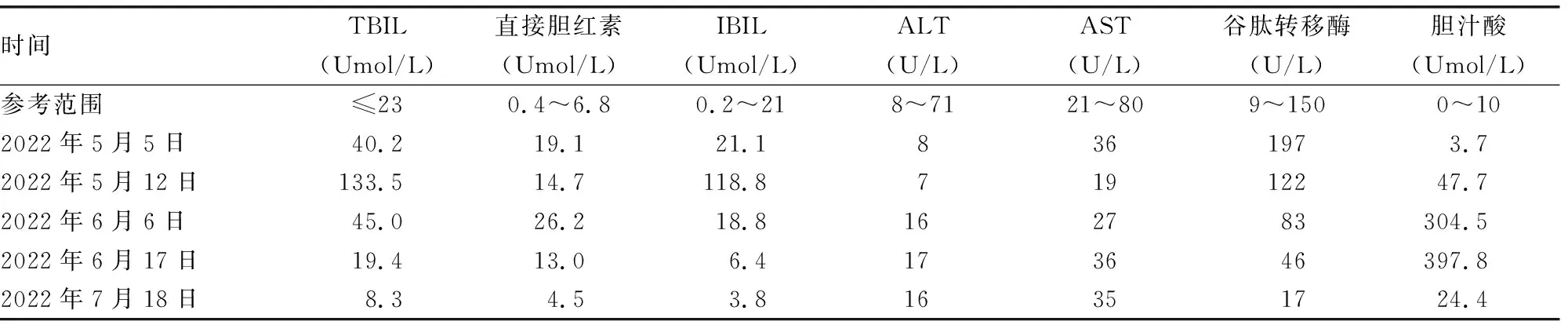
表2 病例2患儿肝功能结果
2 讨 论
新生儿NTCP缺陷病是一种常染色体隐性遗传病, NTCP是由位于人类14号染色体的SLC10A1基因编码,定位于肝细胞的基底外侧膜,肝脏对胆盐的摄取是由钠依赖和非钠依赖性转运蛋白介导的[3];NTCP是一种钠依赖性转运体,主要负责正常生理条件下胆汁酸的转运,以维持胆汁盐不间断的肠肝循环,其缺乏会导致循环胆汁酸水平升高[1,4-5]。在已报道的NTCP缺陷病患者中SLC10A1基因变异c.8000>T(p.Ser267Phe)最为常见,SLC10A1变异在东亚人群中普遍存在,我国发生此病的患者中的等位基因频率达95.5%左右[1,6]。
NTCP缺陷病患者可表现为持续且顽固的高胆汁酸及脂溶性维生素的缺乏。据文献报道,p.Ser267基因突变会导致胆汁酸摄取功能几乎完全丧失[7-9]。本研究病例1患儿的临床表现与此一致,而脂溶性维生素的缺乏可能是与肠道中胆盐的供应减少或饮食有关[10]。脂溶性维生素缺乏可能会影响凝血功能及骨密度下降,故需及时补充所缺乏的脂溶性维生素。还有部分患儿表现为轻微的生长、发育受限。2017年VAN HERPE等[11]报道的随访患者认知发育正常,生长未受限。同时部分患儿还存在反复发作的荨麻疹样皮疹。因目前样本数较少,暂时对于患儿的长期预后,包括生长及发育、肝功能受损等仍需继续进一步随访。
新生儿NTCP缺陷病的长期后果目前尚不清楚,早产儿与足月儿的临床表现也不尽相同。迄今为止,在已被报道的NTCP突变的患者中尚未见到严重的ALT水平升高[12]。本研究病例1患儿为早产儿,病例2患儿为足月儿,足月儿仅2个月左右胆汁酸就逐渐恢复正常,而早产儿1岁时胆汁酸才逐渐恢复正常,是否与体内NTCP酶的活性不同有关。最近一项使用高效液相色谱分析的研究表明,1岁前的血清总胆汁盐水平比1岁后高90%[1,13],故本研究2例患儿暂未口服促胆汁酸排出的药物,以定期复查为主。
NTCP缺陷病患者胆汁酸水平可能不会无法控制地升高,可能会随着年龄的增长而下降,胆汁酸水平升高的具体原因不明,一部分原因可能是生理性胆汁淤积,这会导致正常新生儿和婴儿由于肝功能不成熟而呈现暂时的高胆汁酸水平。新生儿生理性胆汁淤积的可能原因是与NTCP的糖基化程度不同有关,出生后NTCP在人类肝细胞中完成NTCP糖基化大约需要1年。本研究2例患儿TBIL、IBIL、ALT、AST水平均未发现升高,尚未发现严重的肝功能受损。有学者对小鼠NTCP进行基因敲除却没有导致胆汁酸代谢紊乱,考虑可能是其他替代途径如OATPs 和微粒体环氧化物水解酶对胆汁酸摄取的代偿作用[4]。因此,当NTCP功能失调可能引起OATP1B1和OATP1B3的代偿性重吸收,导致肝细胞胆色素沉积,导致胆红素尤其是直接胆红素的继发性升高,本研究2例患儿暂未发现明显的直接胆红素升高。
综上所述,NTCP缺陷病的长期预后仍有待于进一步研究。33.3%的患儿因黄疸迁延不退而就诊,但因大多数黄疸呈自限性,且未并发其他症状,故仍有部分病例被漏诊或误诊,NTCP缺乏发生率为0.64%[5]。因此,对皮肤反复黄染的患儿还应及时完善胆汁酸检查,必要时完善基因检查,做到不遗漏及不过度治疗 。本研究病例1早产患儿以反复黄疸就诊,病例2足月儿因常规复查发现该病,2例患儿目前预后均较好,对NTCP缺陷病的长期预后仍需随访更多此类疾病的患儿。
猜你喜欢
种子(2021年3期)2021-04-12 01:42:22
浙江中医杂志(2019年3期)2019-01-05 23:46:19
祝您健康·文摘版(2018年6期)2018-10-21 18:28:47
猪业科学(2018年8期)2018-09-28 01:27:22
中成药(2018年7期)2018-08-04 06:04:22
护士进修杂志(2017年2期)2017-02-16 06:40:18
蚌埠医学院学报(2016年7期)2016-09-01 06:44:13
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
