
CO2诱导肼类燃料低温反应机理研究
2023-06-12 13:37陈睿哲刘永峰毕贵军宋金瓯
火炸药学报 2023年5期
陈睿哲,刘永峰,王 龙,毕贵军,宋金瓯
(1.北京建筑大学 北京市建筑安全监测工程技术研究中心,北京 102627; 2.四川大学 化学工程学院,成都 610065;3.新加坡科技研究局 制造技术研究院,新加坡 637662;4.天津大学 先进内燃动力全国重点实验室,天津 300072)
引 言
肼类燃料主要包括肼(N2H4)、甲基肼(MMH)和偏二甲肼(UDMH)及其混合物,因其具有热值高、比冲高、易贮存等优点,通常与N2O4组成双组元自燃推进剂,广泛应用于火箭推进系统。然而,肼类燃料具有较强的挥发性、致癌性和爆炸性,其作为强还原剂还会与推进剂配方中如KMnO4、Ca(ClO)2等氧化剂发生剧烈反应产生剧毒物质,使得其生产、运输、贮存和加注过程都极具危险性。针对上述问题,一些学者提出了凝胶法来解决上述问题[1]。
凝胶法是指用凝胶剂、含能颗粒及表面活性剂等使燃料凝胶化,其在储藏时为稳定固态,在加热、加压及剪切等条件下可转变为液态[2],通过管道输送到火箭发动机实现快速雾化、着火与燃烧[3]。用于肼类燃料的凝胶剂主要为高分子聚合物凝胶剂[4],包括聚异丙基丙烯酰胺(PNIPAM)[5]、壳聚糖[6]和羟丙基纤维素(HPC)[7]等。但高分子聚合物凝胶剂制得的凝胶推进剂的黏度较大,不利于加注和点火,燃烧效率也较低,而使用CO2诱导方法所制得的凝胶推进剂大大缓解了这一问题。Allan等[8]最早将此方法使用到肼类燃料及其衍生物的凝胶制备中,发现该方法不仅降低了燃料的挥发性,而且改善了黏度和能量分布。此后大量学者对此方法进行了相关实验研究,Nam等[9]研究了N2H4衍生物乙二胺(EDA)在333~473 K、0.3~10 MPa环境下与CO2的反应,发现在较低温度下更容易形成凝胶。Ethier等[10]在298 K的常压环境下在不同溶剂中采用CO2与多种苄基胺反应成功制备了凝胶,并且发现随温度升高凝胶体系逐渐被破坏,温度升至约323 K时凝胶开始恢复为苄基胺。Pollet等[11]研究了在273 K温度下CO2诱导N2H4、MMH、UDMH和EDA制备凝胶的反应,发现UDMH与CO2反应得到了一种潮湿的白色固体,而其他3种物质均成功制得凝胶。与Ethier的实验现象类似,他发现加热或在凝胶中添加水/乙酸乙酯都可以实现燃料的恢复。为确定CO2诱导肼类燃料在低温下的反应产物,Byeongno等[12]通过X射线衍射分析的方法测试了N2H4与CO2在353 K、10 MPa环境下反应得到的稳定凝胶,判断其是由以N2H4CO2为单体聚合而成的高分子化合物。随后,Byeongno等[13]进一步实验分析了N2H4与CO2反应产生的聚合物的单体,判断其主要为单加合物NH2NHCO2。目前对于CO2诱导肼类燃料的低温反应大多基于实验现象来分析,缺乏通过微观本质来分析肼类燃料与CO2低温反应机理的研究。
本研究采用量子化学计算的方法对N2H4、MMH和UDMH三种肼类燃料在不同温度下与CO2反应的过程进行模拟,分别对其反应活性位点、反应势能面和反应速率进行探讨,以探明CO2诱导肼类燃料的低温反应机理,进而为新型肼类凝胶推进剂的设计与研发提供理论支持与参考。
1 计算方法
1.1 反应位点预测
通过分子动力学模拟(反应力场)、基于量子化学计算不同反应位点的反应通道和基于反应物自身性质预测(波函数分析)的方法[14]可以准确预测反应位点。其中,采用分子动力学模拟的方法计算时间较长,因此用来解决实际问题有一定困难。基于量子化学进行计算主要是通过反应势垒来检测不同位点上反应的难易程度,但这种研究方法十分复杂。而基于反应物自身性质来对反应位点进行预测可以直接考察不同位点发生反应的难易,快捷方便,因此本研究采用此类方法进行计算。基于反应物自身性质来对反应位点进行预测,根据不同原理包括静电势(ESP)、平均局部离子化能(ALIE)、前线分子轨道理论(Frontier Molecular Orbital, FMO)、福井函数(FuKui)、双描述符(Dual Descriptor, DD)等。
其中ALIE[15]是指从分子构成系统的某点移开一个电子所需要的能量,其极小值点表示电子排布最密处,即与亲电分子或自由基反应最有利处。因此所研究的分子表面上ALIE越低,电子束缚越弱,活性越强,就越容易发生反应,其表达式为:
(1)
式中:ρ为总密度,e/m3;ρi为第i个分子轨道所对应的电子密度,e/m3;εi为第i个分子轨道的能量,eV。
考虑到仅通过ALIE来预测反应位点是不可靠的,并且福井函数/双描述符与ALIE这两种方法之间互不矛盾,因此将ALIE和福井函数/双描述符结合进行联合分析可以更好地预测3种肼类燃料分子的活性位点。福井函数[16]描述了当分子构成的体系电子数发生变化时各处电子密度的变化程度,即系统化学势对外部势能的响应,定义式为:
(2)
式中:N为当前体系的电子数;μ为体系化学势,kJ/mol;ν(r)为原子核对电子产生的吸引势,V;ρ(r)为该位点的电子密度,e/m3。
电子密度相对于N的偏导数在N为整数时不连续,应采用有限差分近似其左右导数与二者平均值,则福井函数分为三类(f-、f+、f0),可分别预测亲电、亲核、自由基反应区域,即:
f-(r)=ρN(r)-ρN-1(r)≈ρHOMO(r)
(3)
f+(r)=ρN+1(r)-ρN(r)≈ρLUMO(r)
(4)
(5)
式中:ρN、ρN+1和ρN-1分别代表体系在原始状态(N电子)、结合一个电子状态(N+1电子)和电离掉一个电子状态(N-1电子)下的电子密度。
双描述符与福井函数有着密切的关系,其近似值可通过f+减去f-计算,数值越负的区域越可能是亲电位点,数值越正越可能是亲核位点。为了更直观地反映双描述符分布,可绘制双描述符等值面图进行分析,图中正值区域越大处越容易发生亲核反应,负值区域越大处越容易发生亲电反应。
1.2 反应路径搜索与分析
首先,采用杂化密度泛函(B3LYP)[17]和极化基组(6-311G+(d,p))[18]对N2H4、MMH和UDMH分子进行结构优化,并通过波函数信息[14, 19-20]进行分子活性位点分析[16]。然后在B3LYP/6-311G+(d,p)基础上对N2H4、MMH和UDMH与CO2反应路径进行搜索,具体过程为:(1)对过渡态(TS)进行初猜,采用B3LYP/6-311G+(d,p)进行初始结构优化;(2)沿着选定的将要形成或断裂的化学键进行弛豫能扫描并获得势能面,扫描级别为B3LYP/6-311G+(d,p);(3)对扫描势能面最高点的结构进行几何结构优化和振动频率分析,级别为B2PLYP-D3(BJ)/def2tzvp;(4)识别TS为一阶的频率分析临界点,采用内禀反应坐标(IRC)对TS进行正反两个方向的最小能量路径搜索,并优化反应路径中的反应物(Rc)、产物(Pc)、过渡态(TS)及中间体(IM)的几何构型,以获得能量最低最稳定的分子结构,IRC计算级别与过渡态计算级别保持严格一致;(5)对优化后的几何构型进行频率分析,确保所得反应物、产物及中间体频率均为正值,且过渡态有且仅有一个虚频;(6)采用CCSD(T)/aug-cc-pVTZ级别计算Rc、Pc、TS与IM的单点能。
基于频率优化后各个结构的单点能,采用Shermo[21]计算不同温度下反应过程的热力学数据变化,通过频率校正因子消除由于谐振近似和计算级别自身原因带来的系统性误差。根据前人实验研究[9,12-13],本研究选取温度范围为333~473 K,压强为10 MPa。
最后通过KiSThelP[22-23]计算反应速率常数,该程序基于经典过渡态理论并以Wigner方法[24]考虑隧道效应校正进行计算:
(6)
2 结果与讨论
2.1 反应位点
2.1.1 平均局部离子化能
基于B3LYP/6-311G+(d,p)对N2H4、MMH和UDMH进行结构优化,分子结构如图1所示。其中N2H4的键长与Yonezo等[25]通过显微光度计获得的数值非常吻合,且MMH与UDMH的结构参数也与文献值[26-27]基本吻合。
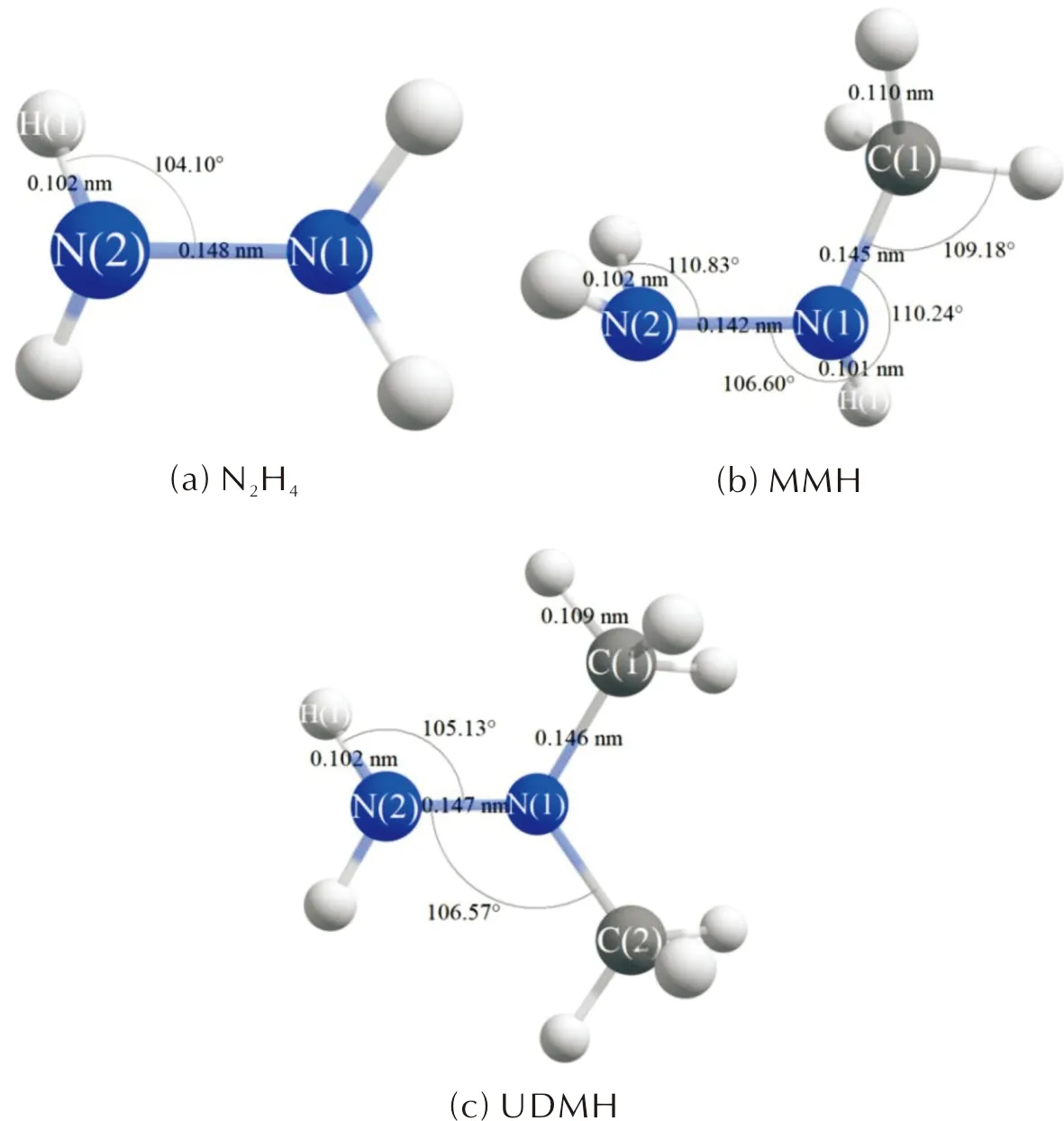
图1 N2H4,MMH和UDMH的几何构型
根据结构优化所得波函数信息利用Multiwfn进行分子活性位点分析,得到N2H4、MMH和UDMH的电子密度为0.000 5 a.u.的等值面,如图2所示,其中红色区域为ALIE数值较大区域,蓝色区域为ALIE数值较小区域,青色圆球则代表区域内的极小值点。

图2 N2H4、MMH和UDMH的ALIE等值面
通过比较可以发现,ALIE较小的区域主要分布在N2H4、MMH和UDMH的N原子附近,N2H4的极(最)小值为9.54 eV,MMH的N(1)比N(2)覆盖蓝色区域大,其中N(1)附近的最小值为9.05 eV,UDMH的N(2)比N(1)覆盖蓝色区域大,其中N(2)附近的最小值为8.70 eV,上述最小值点与赵建铄等[28]得出的结果一致。
2.1.2 福井函数/双描述符
采用Multiwfn程序对N2H4、MMH和UDMH的函数数据进行计算,设置等值面数值为0.01 a.u.,所得双描述符等值面如图3所示。
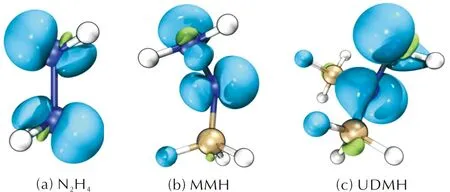
图3 N2H4、MMH和UDMH的双描述符等值面
图3中绿色和蓝色部分分别代表此处双描述符数值为正或为负,可以看出,在N2H4、MMH和UDMH的N(1)和N(2)上蓝色区域较大,即双描述符为负的地方,说明三者的N(1)和N(2)容易被亲电进攻。同时,在MMH和UDMH的C原子部分区域出现了面积较小的绿色区域,即双描述符为正的地方,说明这些区域有被亲核进攻的可能,但反应活性与N原子相比活性较低。
为定量分析N(1)与N(2)的活性强弱,采用轨道权重双描述符定量分析法对二者进行比较,计算结果如图4所示,其中红球和蓝球分别对应ρ=0.01 a.u.等值面上双描述符值的极大点和极小点。可以看出,N2H4的两个N原子处数值最小约为-8.14×10-2eV,MMH的N(1)原子处数值最小约为-7.57×10-2eV,UDMH的N(2)原子处数值最小约为-6.19×10-2eV,表明这些位置是电子最容易被亲电进攻的位置。同时可以发现,其他极值点数值与N原子处数值相比相差较大,因此这些地方不易发生亲电或亲核反应。Nigst等[29]实验测定了甲基取代肼类燃料不同位置对其反应活性的影响,发现甲基取代基可使被取代的N原子亲核性提高,使邻位原子的反应活性降低。而从本研究中对MMH双描述符数值的计算可以看出,与甲基相连的N原子比未与甲基相连的N原子亲核性更强,但对UDMH的分析却相反。
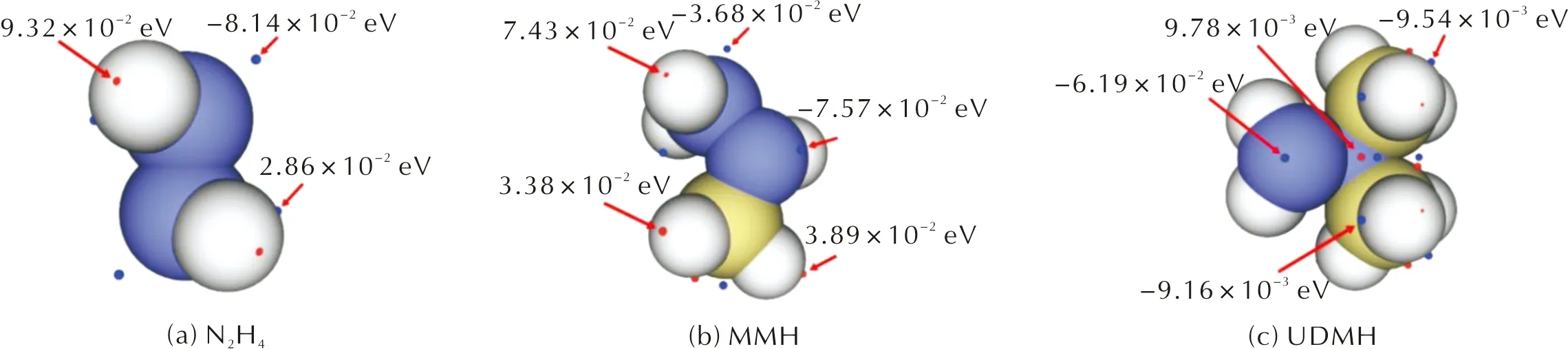
图4 N2H4、MMH和UDMH的双描述符定量分析
由此可见,3种肼类燃料分子的ALIE与福井函数/双描述符的计算结果一致。可以得出,N2H4的反应活性位点为N原子,MMH的反应活性位点为N(1)原子,UDMH的反应活性位点为N(2)原子。其中UDMH的N(2)原子为氨基的N原子,王力等[30]对臭氧与UDMH的氧化反应进行了研究,发现氨基上的氢原子最容易脱离生成自由基(CH3)2NNH·进一步氧化,即氨基处N原子活性最强,本研究的结论很好地解释了这一现象。
2.2 反应路径
根据双描述符等值面图判断3种肼类燃料的活性位点N原子既可被亲电攻击也可被亲核攻击,采用B2PLYP-D3(BJ)/def2tzvp级别方法构建了CO2的C、O原子攻击N2H4、MMH和UDMH的N原子的反应路径,计算所得CO2的O原子攻击N原子的反应路径如图5所示,反应势能面如图6所示。在CO2与N2H4的反应中,CO2一端的C—O双键变为单键,且此处的O原子与N原子连接成键,通过过渡态能垒454.84 kJ/mol形成了TS结构,经过计算,TS存在唯一虚频,且虚频振动方向与预测的成键方向完全一致,因此TS为正确的过渡态结构,其中N—O键长为0.155 nm,C—O单键被拉长0.026 nm,O—C—O键角减小67.18°,C—O—N键角为109.48°。最后TS中C—O键断开,释放了135.21 kJ/mol的能量,N—O键长减小0.022 nm,形成了产物结构。
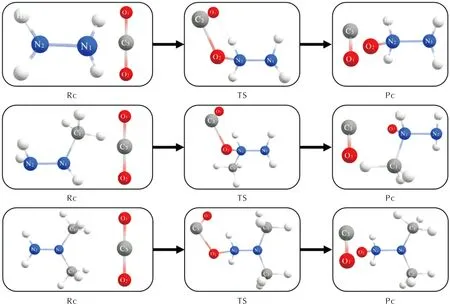
图5 CO2的O原子攻击N2H4、MMH和UDMH的N原子反应路径

图6 CO2的O原子攻击N2H4、MMH和UDMH的N原子反应势能面
在CO2与MMH的反应中,O原子与N(1)原子连接成键,通过过渡态能垒435.57 kJ/mol形成了TS结构,所得TS结构N—O键长为0.155 nm,C—O单键被拉长0.026 nm,O—C—O键角减小66.81°,C—O—N键角为110.24°。最后TS中的C—O键断开形成了产物结构,释放了108.73 kJ/mol的能量,N—O键减小0.022 nm。在CO2与UDMH的反应中,O原子与N(2)原子连接成键,通过过渡态能垒441.67 kJ/mol形成了TS结构,所得TS结构N—O键长为0.155 nm,C—O单键被拉长0.029 nm,O—C—O键角减小68.36°,C—O—N键角为110.02°。最后TS中的C—O键断开形成了产物结构,释放了110.84 kJ/mol的能量,N—O键减小0.021 nm。
CO2的C原子攻击N原子的反应路径如图7所示,反应势能面如图8所示,可以看出,该反应路径最终产物均为肼羧酸结构。在CO2与N2H4的反应中,CO2的C原子首先与N原子连接成键,通过过渡态能垒198.33 kJ/mol形成了TS结构,经验证,TS为正确的过渡态结构,其中C—N键长为0.156 nm,O—C—O键角减小41.66°。然后CO2所连接的N原子上的一个H原子掉落并被CO2上的O原子夺走形成了羧基,释放192.6 kJ/mol的能量形成最终产物,其中O—H键长为0.097 nm,H—O—C键角为105.04°。
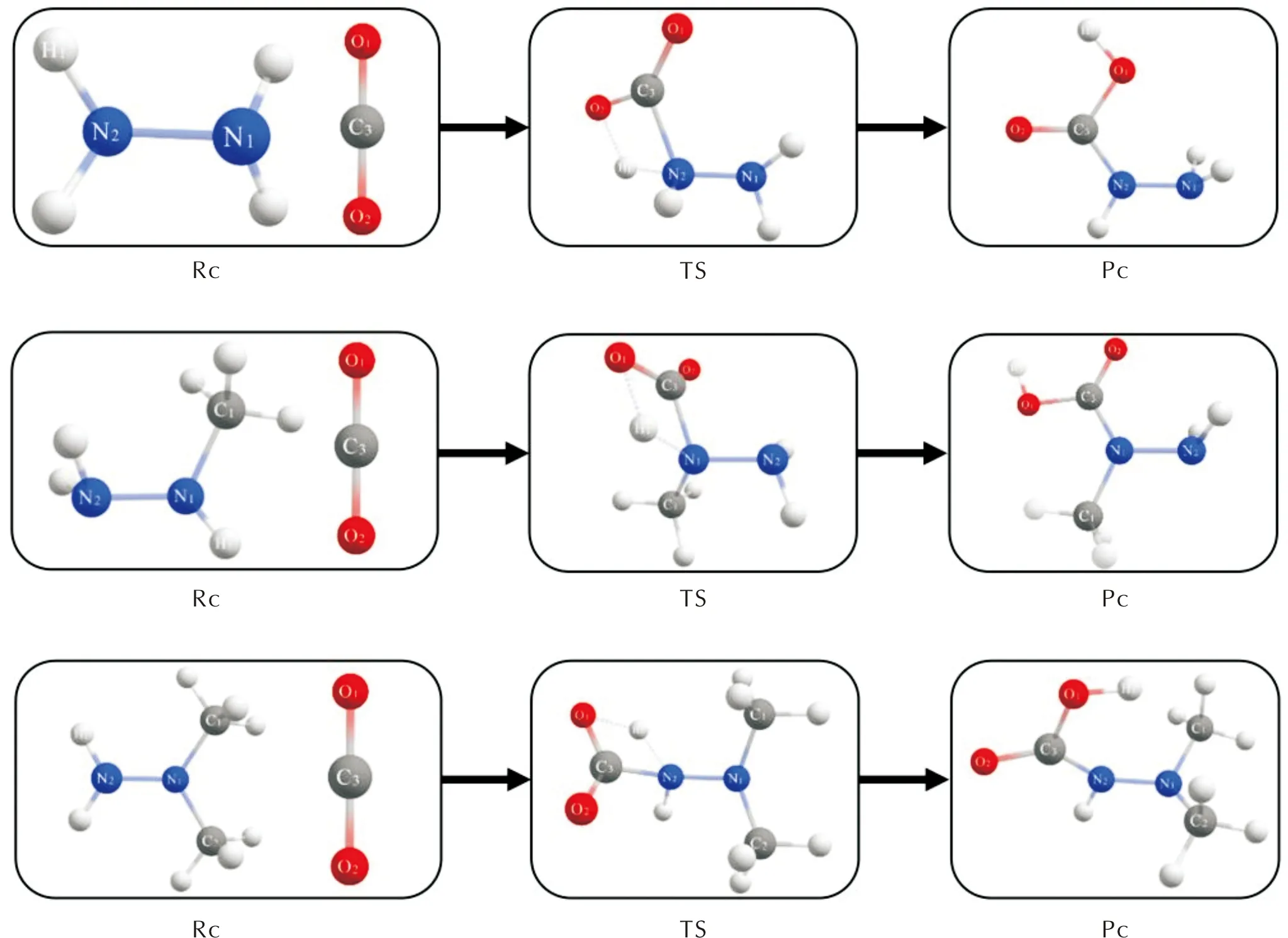
图7 CO2的C原子攻击N2H4、MMH和UDMH的N原子反应路径

图8 CO2的C原子攻击N2H4、MMH和UDMH的N原子反应势能面
Byeongno等[12]对N2H4和CO2反应产生的凝胶通过单晶x射线衍射得到了凝胶结构,其单体与本研究计算所得单体结构一致。在CO2与MMH的反应中,C原子与N(1)原子连接成键,通过过渡态能垒207.24 kJ/mol形成了TS结构,其中C—N键长为0.156 nm,O—C—O键角减小42°。然后CO2所连接的N(1)原子上的一个H原子掉落并被CO2上的O原子夺走形成了羧基,释放193.09 kJ/mol的能量形成最终产物,其中O—H键长为0.097 nm,H—O—C键角为104.79°。在CO2与UDMH的反应中,C原子与N(2)原子连接成键,通过过渡态能垒218.32 kJ/mol形成了TS结构,其中C—N键长为0.158 nm,O—C—O键角减小43°。然后CO2所连接的N(2)原子上的一个H原子掉落并被CO2上的O原子夺走形成了羧基,释放160.51 kJ/mol的能量形成最终产物,其中O—H键长为0.096 nm,H—O—C键角为110.71°。
Pollet等[11]在1.01×105Pa、273 K下进行了使用CO2制备肼类凝胶的实验,发现N2H4和MMH均可制得凝胶,但UDMH与CO2反应却很难得到凝胶。从上述3种肼类燃料与CO2的反应势能面对比中可以看出,在两条反应路径下均呈现同样的规律:CO2与N2H4反应产物与反应物能量差最小,MMH次之,UDMH最大,说明CO2分别与N2H4、MMH和UDMH反应的难度也依次增大。
2.3 反应路径的热力学分析
为进一步探究肼类燃料与CO2低温反应的性质,本研究选取温度范围为333~473 K,压强为10 MPa,分别对反应路径中Rc、TS和Pc的热力学参数包括矫正焓变ΔH、矫正自由能变ΔG和矫正熵变ΔS进行计算,计算结果如图9所示。
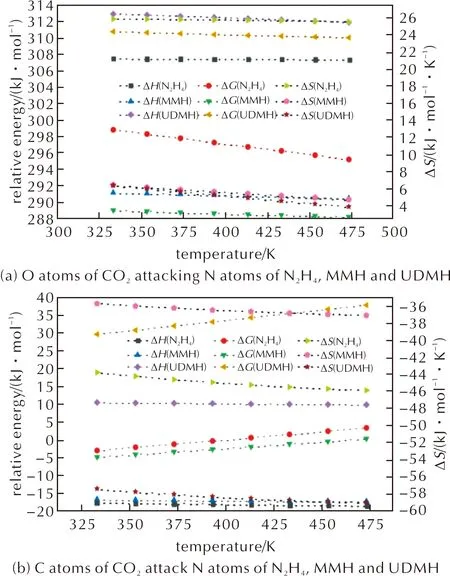
图9 不同温度下CO2与N2H4、MMH和UDMH反应中物质的热力学参数
由图9可以看出,在温度333~473 K范围内,CO2的O原子攻击N2H4、MMH和UDMH的N原子的反应ΔH均呈下降趋势,且随T的不断上升,ΔS均平缓下降,根据ΔG=ΔH-T·ΔS计算所得ΔG不断下降,与图9(a)中所得ΔG的递减趋势一致,即该体系从高能量状态过渡到低能量状态,说明该反应路径符合化学反应动力学。此外,对于3种肼类燃料反应的ΔG均为正,说明在此温度范围内此条反应不能自发进行,并且反应的ΔH与ΔS均为正,说明此条反应路径只能在高温下自发进行。而根据现有研究中CO2诱导法的反应条件[9,12-13],说明此条反应路径不是CO2诱导法制备肼类凝胶推进剂的反应。对比3种肼类燃料在同一温度下与CO2反应的ΔH,MMH的ΔH最小,N2H4的ΔH次之,UDMH的ΔH最大,意味着UDMH的反应过程吸热最多,反应所需的温度条件最高。
如图9(b)所示,在温度333~473 K范围内,CO2的C原子攻击N2H4、MMH和UDMH的N原子的反应ΔH和ΔS随温度T的上升均呈缓慢下降趋势,而ΔG均为递增趋势。其中N2H4与CO2反应的ΔG从-2.97 kJ/mol上升到3.33 kJ/mol,约在400 K时ΔG由负值变为正值,说明在333~400 K范围内,此反应可以自发进行,且根据ΔG的递增趋势,不难推断出在低于333 K的温度下,该反应仍然自发进行。而在400~473 K范围内,此反应不可自发进行。同时,反应的ΔH和ΔS均为负值,也印证了该反应在低温下自发进行。Lee等[31]实验测定了N2H4对CO2的吸附作用,结果表明在温度313~393 K范围内N2H4会与CO2反应生成无色凝胶,而加热到393 K以上温度时,凝胶结构会被破坏,使得CO2从产物中分离,这与本研究所预测反应路径的理论计算结果一致。MMH与CO2反应的ΔG从-4.80 kJ/mol上升到0.31 kJ/mol,约在460 K时ΔG由负值变为正值,说明在333~460 K范围内,此反应可以自发进行,且根据ΔG的递增趋势,推断在低于333 K的温度下,该反应仍然自发进行。而在460~473 K范围内,此反应不可自发进行,且反应的ΔH和ΔS始终为负值,也印证了该反应在低温下自发进行。UDMH与CO2反应的ΔG从49.62 kJ/mol上升到57.78 kJ/mol,始终为正值,说明在333~473 K范围内,此反应均不可自发进行,且反应的ΔH始终为正值,ΔS始终为负值,表明该反应在任何温度下均不可自发进行。Pollet等[11]在压力1 atm、温度273 K下使用CO2和UDMH制备凝胶但未成功,此实验现象在本研究计算结果中也得到了论证。对比3种肼类燃料在同一温度下与CO2反应的ΔH,UDMH的ΔH最大,MMH的ΔH次之,N2H4的ΔH最小,意味着N2H4的反应过程吸热最少,反应更利于在低温下进行。
2.4 反应路径的动力学分析
通过KiSThelP程序计算所得的不同温度下N2H4、MMH和UDMH与CO2的反应速率常数见表1。

表1 不同温度下CO2与N2H4、MMH和UDMH的反应速率常数
由表1可以看出,3种肼类燃料与CO2的反应速率常数均随着温度的升高而增大,并且在N2H4和MMH与CO2的低温正向反应中,MMH的反应速率常数比N2H4的反应速率常数大,说明在同一温度下CO2与MMH的反应速度更快。Pollet等[11]使用N2H4和MMH吸收CO2的实验表明,在1.01×105Pa、273 K下同一时间内等量MMH所吸收的CO2大约是N2H4的4~5倍,这从实验上为上述结论提供了支撑。此外,在3种肼类燃料与CO2的逆向反应中,N2H4、MMH和UDMH的反应速率常数依次增大,即在同一温度下UDMH更容易从其与CO2所构成的凝胶单体中恢复。
3 结 论
(1)N2H4的反应活性位点为N原子,MMH的反应活性位点为与甲基相连的N原子,UDMH的反应活性位点为氨基中的N原子。
(2)肼类燃料与CO2的反应主要存在两条路径:一是CO2的O原子攻击肼类燃料的N原子,其主要产物为N2H4O/CH6N2O/C2H8N2O,该反应在低温下不会发生;二是CO2的C原子攻击肼类燃料的N原子,其主要产物为肼羧酸,该反应为CO2诱导肼类燃料低温反应形成凝胶单体的主要反应。
(3)在10 MPa的环境压力下,N2H4与CO2反应形成凝胶单体的最高温度约为400 K,与MMH和UDMH相比,其反应所需热量最小,在393 K下为17.97 kJ/mol。MMH与CO2反应形成凝胶单体的最高温度约为460 K,而UDMH很难与CO2反应形成凝胶单体。
(4)肼类燃料形成凝胶单体的速率和肼类燃料从凝胶单体中恢复的速率均随温度的升高而增大,MMH与CO2反应形成凝胶单体的速度最快,在10 MPa、453 K环境下的反应速率常数为3.51×10-8L/(s·mol),其从凝胶单体中恢复的速度也最快,在10 MPa、473 K环境下反应速率常数为2.14×10-5L/(s·mol)。
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
测绘学报(2022年12期)2022-02-13
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
大学化学(2021年8期)2021-09-26
计算机应用与软件(2020年6期)2020-06-16
数字通信世界(2018年1期)2018-04-18
电脑知识与技术(2018年3期)2018-03-21
测绘科学与工程(2017年5期)2017-05-07
