
层状含能材料设计方法研究进展
2023-06-12 13:37何晓凯曹意林刘英哲
火炸药学报 2023年5期
何晓凯,曹意林,刘英哲
(1.西安近代化学研究所,陕西 西安 710065;2.西安市液晶与有机光电转换材料重点实验室,陕西 西安 710065)
引 言
含能材料是一类含有爆炸性基团或含有氧化剂和可燃物、能独立进行化学反应并输出能量的化合物或混合物,是军用炸药、发射药和火箭推进剂配方的重要组成部分(本文中如无特殊说明,含能材料的内涵仅为化合物)。能量和感度是含能材料最重要的性能标度,但二者之间通常具有一定矛盾,即随着能量的升高,感度往往呈现下降趋势。因此,开发能够平衡能量和感度关系的新型含能材料是当前的难点问题[1-10]。
含能材料能量和安全性能是由多个结构尺度因素共同决定的。在分子层面,化学储能键往往是最先发生分解反应的键,因此导致能量和安全性能间的矛盾不可避免的出现于分子层面[8]。在晶体层面,通过特定的晶体堆积模式能够一定程度上缓解能量和安全性能间的矛盾。例如,常温常压下CL-20存在3种单质晶型(β、γ和ε型),其中ε-CL-20因其最高的密度(2.04 g/cm3)和堆积系数(0.77)[11],表现出优于其他晶型的爆轰性能和感度性能。因此,低感高能含能材料的设计在分子层面较难实现,需面向晶体尺度设计含能材料。
层状含能材料是一类由平面有机含能分子通过氢键、π-π堆垛等分子间作用形成的具有层状堆积特性的晶体含能材料,是极具潜力的低感高能材料[12-21]。主要原因有3个方面:一是层状堆积含能晶体通常具有较高的堆积系数,能够提升晶体密度、减少自由体积,有助于改善爆轰性能与撞击感度[22-24]。二是层与层之间还可通过滑移来缓冲外界的机械刺激。例如,TATB是典型的层状含能材料(见图1),具有极低的撞击感度(撞击能>60 J),但爆速仅为8 100 m/s左右,能量水平相对较低[25]。三是层状含能材料的热稳定性较好。本课题组[23]从1 000多个实验测量的硝基晶体中筛选出了5种具有层状堆积特点的多晶型化合物,通过计算发现,同一晶型中层状堆积结构的初始热分解反应表观活化能最高。因此,层状含能材料具有天然的机械刺激不敏感性,若在结构设计中辅以能量提升,便能够在晶体层面上较好地平衡能量与机械感度。

图1 典型层状含能材料TATB结构示意图
鉴于分析分子间复杂相互作用和精确预测晶体结构的困难,层状含能晶体的理性设计仍然是一个挑战[26-29]。当前,层状含能材料的设计主要集中在分子层面,例如不同骨架(如苯和噁二唑)与基团(如硝基、氨基和叠氮基)之间的组合设计[30-34]。在大多数情况下,分子的微小修饰会改变晶体的结构和性能(特别是机械感度)[35],从而增加了构建层状含能晶体的难度。
因此,层状含能材料的发展较为缓慢,在已经发现的含能材料中,层状含能材料的种类屈指可数。为了加快层状含能材料的创制,亟需深入理解含能晶体中分子间的相互作用,并创新含能材料设计理论与方法。为此,本文综述了近年来层状含能材料设计方法研究进展,以期为相关研究人员提供参考借鉴。
1 层状含能材料的现有设计策略
近年来,国内外成功获得了若干新型层状含能材料。其中,具有代表性的结构如图2所示。这些含能材料具有完美的层状堆积特征,均表现出了较低的撞击感度(见表1)。值得一提的是,NAPTO具有与HMX相近的爆轰性能,但撞击感度却显著降低,达到了能量与感度的较好平衡[14]。与传统含能材料相比,在设计层状含能材料时,除了要考虑平面含能分子的结构组成之外,还要考虑层状堆积结构的精准构筑。目前,层状含能材料的设计思路重点围绕堆积结构特点开展,其设计策略可以分为三类:一是基于经验,二是基于计算,三是基于机器学习(见图3)。
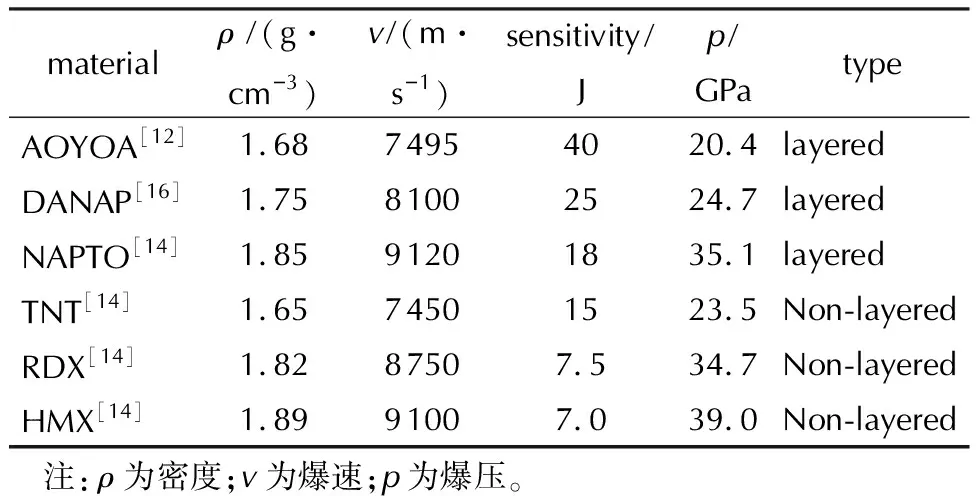
表1 近年来合成出的层状含能材料代表的综合性能
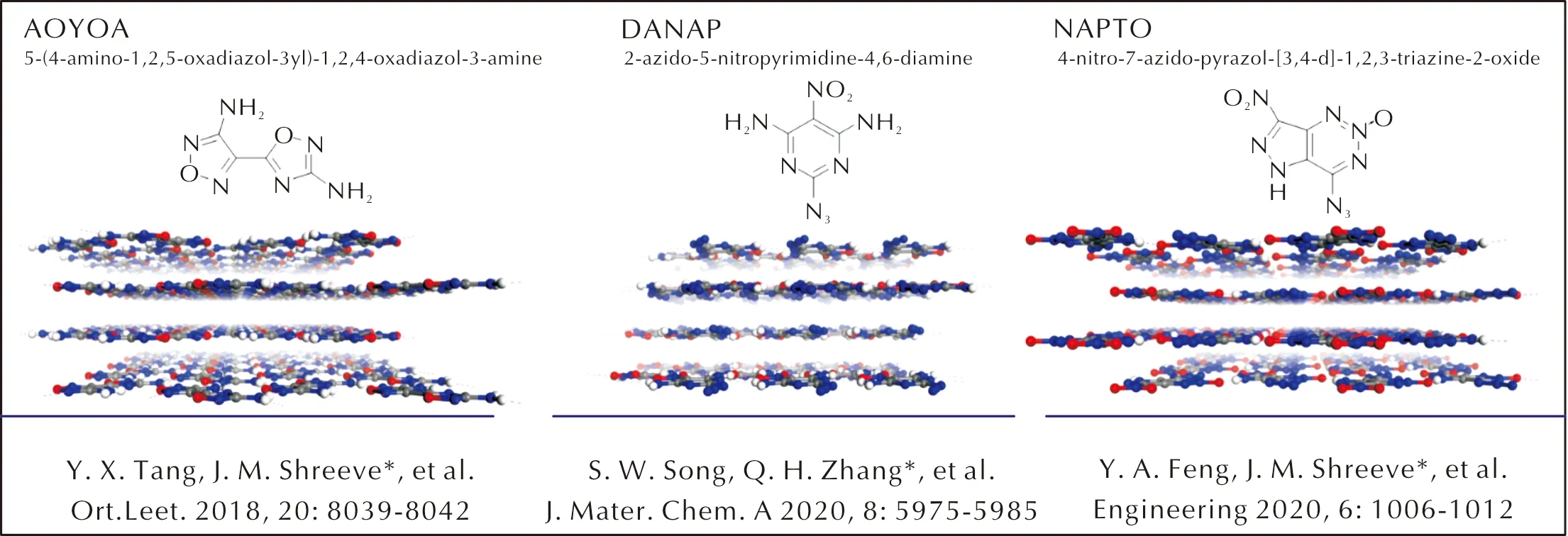
图2 近年来合成的典型层状含能材料
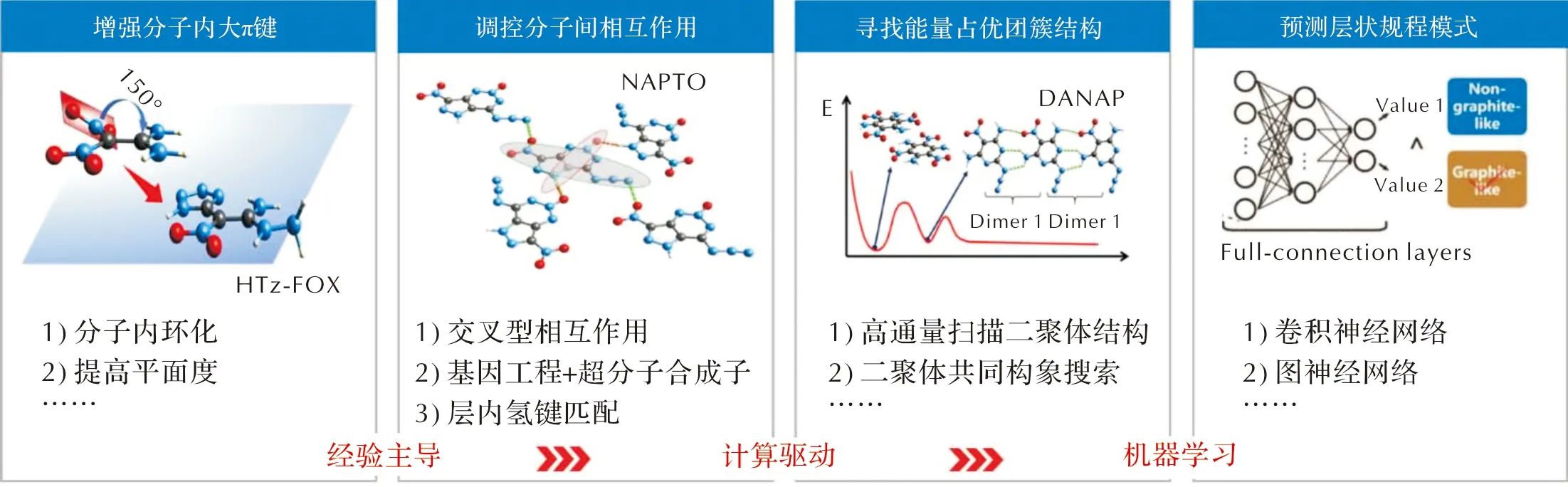
图3 层状含能材料设计策略汇总
1.1 经验驱动设计模式
通过分析一些已有层状含能晶体的堆积规律[36-37],获得可能形成层状堆积的影响因素,比如分子平面性、空间对称性、氢键/π-π堆垛作用等,把这种依赖规律性的方法归结为基于经验驱动的设计策略,主要分为增强分子内大π键[12-13,19,38-39]、调控分子间相互作用[14,18,25,40-44]。
制备层状材料的关键是合成平面分子,因FOX-7中同C原子上两个NO2间的空间位阻破坏了分子的平面性,导致FOX-7具有波浪状晶体结构,并不是较为平整的层状结构。将FOX-7分子中的一个NH2和一个NO2基团分别替换为联胺和四唑环,形成的平面分子HTz-FOX具有层状晶体结构,其感度(IS=10 J)和能量性能(D=8 883 m/s)优于RDX[19,38,45]。
内环化反应也是一种提高分子平面性的重要方式。如图4(a)所示,Tang等[12]利用碘苯二乙酸酯在二甲基甲酰胺溶液中氧化分子4-氨基-N-(二氨基乙烯)-1,2,5-噁二唑-3-甲酰胺,得到化合物5-(4-氨基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-3-胺,反应前后分子内参与共轭的原子比例增加,共轭效应增强,最终晶体呈现层状堆积模式,表现出优于TNT的能量(D=7 495 m/s)和安全性能(IS>40 J)。Yang的团队[13]通过分子内环化反应同时制备了两种层状含能化合物(见图4(b)),其中化合物4,6-二硝基-(1H-四唑-基)-1H-吲唑表现出较好的爆轰性能(D=8 518 m/s)和安全性能(Td=316℃、IS=27 J)。
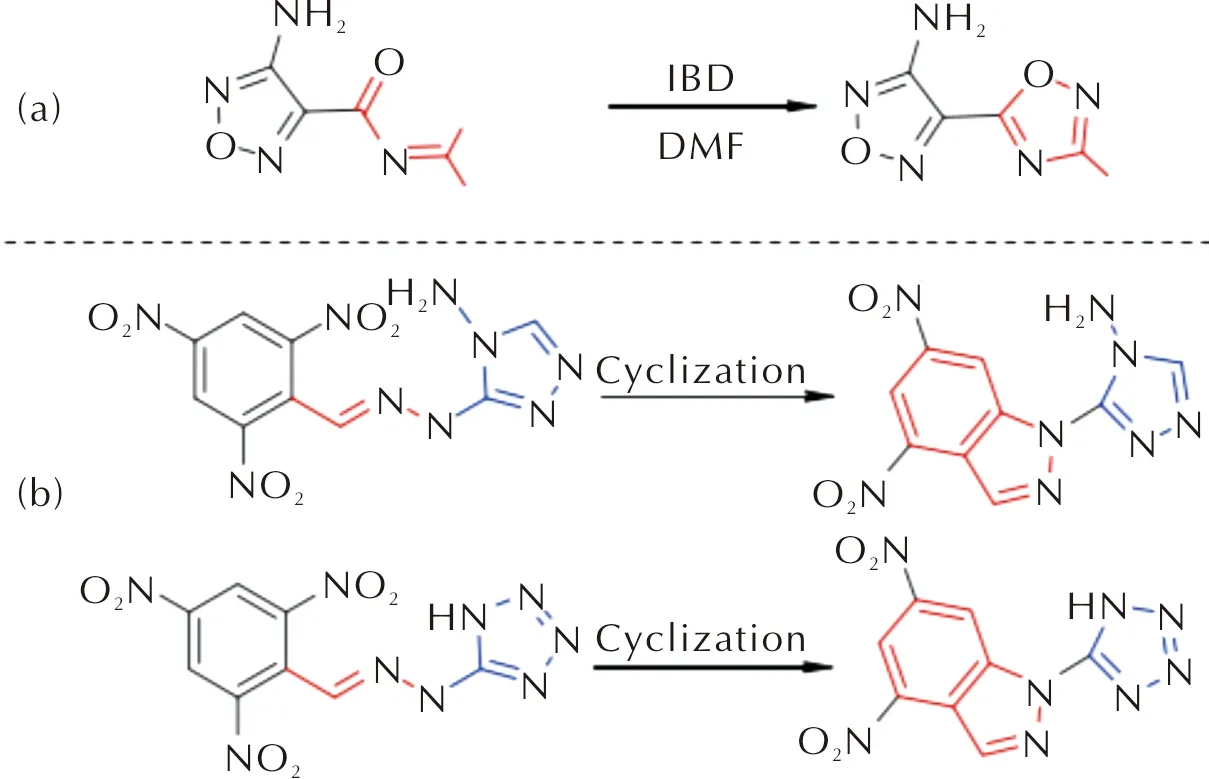
图4 通过环化反应合成层状含能材料
在调控分子间相互作用方面,Feng等[14]利用HBs(氢键)和偶极-偶极相互作用,创造了一种很有前途的含能材料NAPTO。NAPTO具有超长二维层状结构(层间π-π紧密堆积,层间距为2.883 Å),还具有较高的爆轰性能(D=9.12 km/s,P=35.1 GPa)、良好的热稳定性(203.3℃)和良好的对外界刺激的低机械感度(IS=18.0 J,FS=325 N,EDS: 0.32 J)。NAPTO的二维层状结构可以有效缓冲外界刺激,从而平衡了NAPTO的高能量和良好的机械感度。
层内HBs和层间π-π作用是层状含能晶体堆积结构的主要构成要素。Shreeve等[40]提出了一种“层内氢键配对方法”的快速且简单的策略,通过逐层堆叠来组织含能分子,从而获得具有目标特性可调的含能材料。同时,该团队[42]还报道了一种合成3-氨基-5-硝基-1,2,4-噁二唑的有效方法,其硝基和氨基在同一个基团上。单晶x射线结构显示了层状氢键对以及水分子的存在。此外,Feng等[18]提出了一种氢键“受体-供体分离”的新策略,通过定向氢键来控制含能分子的逐层堆积:即氢键的供体和受体位于不同的含能片段,其中至少有一个具有共轭平面结构,可使含能片段在二维平面上无限扩展,形成目标层状结构。
北京理工大学Pang等[46]提出了榫卯策略,以作为“榫”的无环化合物(二硝基脲的氨基胍盐)和与之匹配的对应化合物(卯)为结合体,以氢键作用为结合力,构建了一个二维的氢键环骨架,从而实现了无环化合物的层状堆积。并通过与之对应的W型堆积结构进行比较,发现稳定性显著提高。
1.2 化学理论计算驱动设计模式
利用理论计算方法对含能分子二聚体、团簇甚至晶体堆积进行能量计算或结构预测,可以从微观角度推断层状结构形成的可能性,从而为实验合成提供目标化合物,这种设计思路称为计算驱动设计模式。由于含能分子间的作用较弱,使得分子堆积的结构种类较多,通常需要较多的计算耗时。
中物院Zhang等[47]高通量计算了1,3,5-三硝基苯及其衍生物分子二聚体的相互作用能,尝试利用热力学稳定的二聚体构型关联对应晶体结构的堆积模式。结果发现,平行堆积的二聚体在热力学上是最稳定的,其次是T型、共面型和交叉型。Li[48]利用此类方法,在设计出的一系列富氮稠环分子中挑选出数种性能优异的层状化合物候选分子。
Zhang的团队[16]提出了一种“共面构型搜索”和“点-链-面”分子间自组装的方法(见图5)来设计和合成层状含能晶体。其具体原理为:在二聚体中的分子共面排列情况下,搜寻能量最低的二聚体用于组装成分子“链”,并进一步组装成分子“面”,其中每一步的组装都需要判断其构型是否合理,能量是否最低。最终,贯通了从分子、二聚体、分子“链”到分子“面”的组装,成功从426个分子中筛选出层状化合物DADNP(2-叠氮-5-硝基嘧啶-4,6-二胺)。
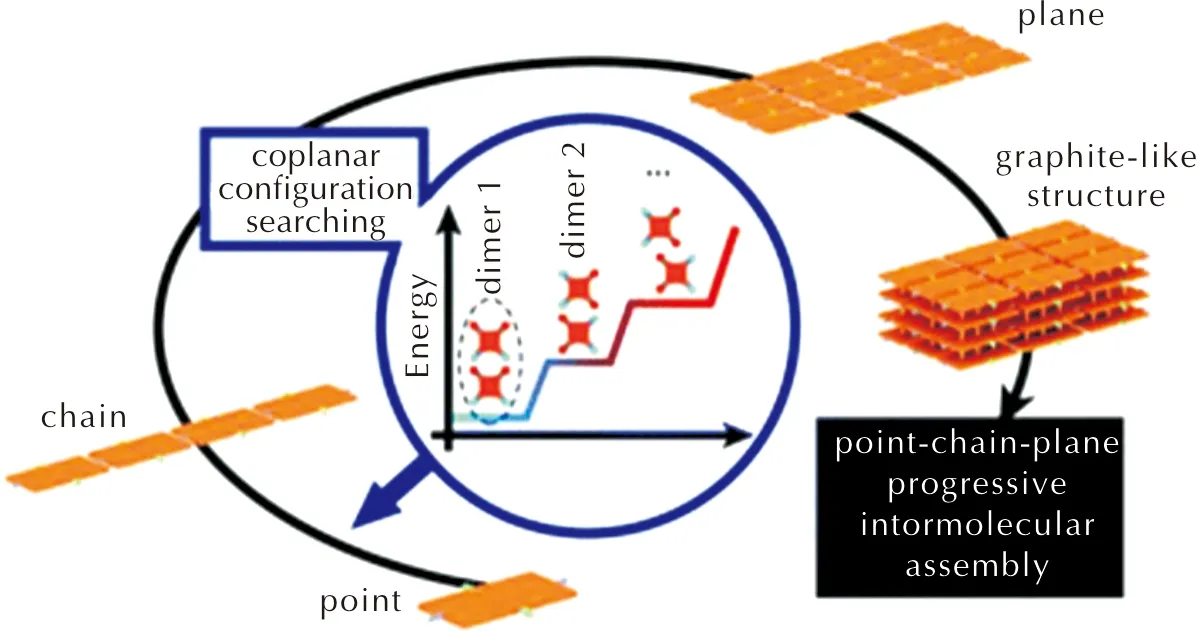
图5 “共面构型搜索”和“点-链-面”分子间自组装的方法[16]
本课题组[49]提出了一种从分子到晶体层次的多层级结构设计策略(见图6)。受到实验中已知的具有特定NH2…NO2分子间基序的高能晶体的启发,通过系统分析结构/组成-性能关系,建立了一套基于H2N-C-C-NO2部分的设计和筛选潜在目标分子的识别模式。随后,利用进化算法USPEX结合最近开发的含能分子可极化力场,对可能的晶体结构进行了精确搜索。理论结果表明,B1(4,4′-二氨基-3,3′-二硝基-[5,5′-二异噁唑]2,2′-二氧化)和C2[(E)-3,3′-(重氮-1,2-二基)双(4-氨基-5硝基异噁唑-2-氧化物)]是具有良好机械感度(优于TNT)和爆轰性能(优于FOX-7)的先进层状含能材料的候选物。
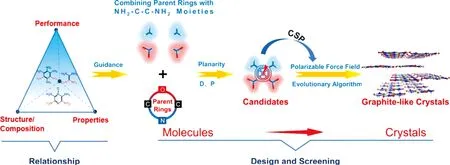
图6 从分子到晶体层次的多层级结构设计策略[49]
上述方法在设计层状含能晶体中取得了较大进展,但平面分子与堆积模式间的关联尚未建立,特别是在高通量设计和筛选分子时,无法快速判断其晶体结构是否为层状堆积结构。因此,建立分子结构和层状堆积模式间的关联,对于设计特定堆积结构的含能晶体具有重要指导意义。
当前,在其他领域的简单体系中已建立了平面分子和堆积模式间的关联。例如,在有机半导体中[51-55],研究发现当并五苯类的分子表面静电势都为正值时,对应晶体为人字形堆积结构,其O—、N—、F—取代物分子中因具有HBs的受体单元,从而表现出层状或交叉状堆积结构。Zhang等[37]通过研究10种分子具有D2h或D3h对称结构并且晶体为层状堆积的化合物,提出了一种基于强氢键和高对称分子准则设计层状晶体结构的策略。此外,Han等[55]开发的“Autopack”软件也被用于自动识别平面分子的堆积模式,并在大样本空间中研究化学组成与堆积模式间的关系。
如何准确地预测层状晶体结构是基于计算驱动的重要研究内容,但较为困难[26]。原因主要有:分子在高维空间中的不同排列方式导致晶体结构样本数量庞大、缺少准确晶格能排序的计算方法以及无法考虑实际结晶过程中的动力学因素等。此外,在大多数的晶体结构预测方法中,往往难以兼顾计算速度和计算精度。
基于高精度分子力场预测晶体结构是一种能够兼顾计算速度与计算精度的有效途径。Rice等[56]开发了Sorescu-Rice-Thompson(SRT)力场,并用以预测174个CHON类晶体结构,其中成功预测出149个晶体,但是SRT力场无法准确描述CL-20的分子间相互作用。Pakhnova等[57]利用反应力场ReaxFF和进化算法USPEX预测了PETN、TATB、HMX、TNT、CL-20以及它们的共晶结构,其中只成功预测出PETN、TATB和HMX的晶体结构。
Neumann[58]提出了一种针对每个分子所建立的定制力场(TalilorMade Force Field,TMFF),可成功用于预测多种化合物的晶体结构。Podeszwa等[59]基于对称匹配微扰理论(symmetry adapted perturbation theory)计算RDX二聚体的结合能用于拟合分子间力场,其拟合出的力场成功预测出RDX晶体结构。Xue[60]针对每个分子建立专属的力场(OPLS-AA),并成功预测出3种CL-20晶型(ε-、β-和γ-)。然而,上述过程涉及到繁琐的二聚体大量抽样与计算过程以及力场参数优化过程。
本课题组采用AMOEBA可极化力场框架,基于第一性原理计算结果,开发了含能材料高精度可极化力场(EMS-PFF)。与COMPASS、DREIDING等经典力场相比,EMS-PFF具有更高的计算精度[61]。结合先进的进化算法,对9种典型硝基平面分子的堆积模式进行了预测。如图7所示,其中8种硝基晶体的堆积模式能够被准确预测出来,说明EMS-PFF结合进化算法具有较高的预测精度[49],可以适用于层状含能材料的晶体结构预测。
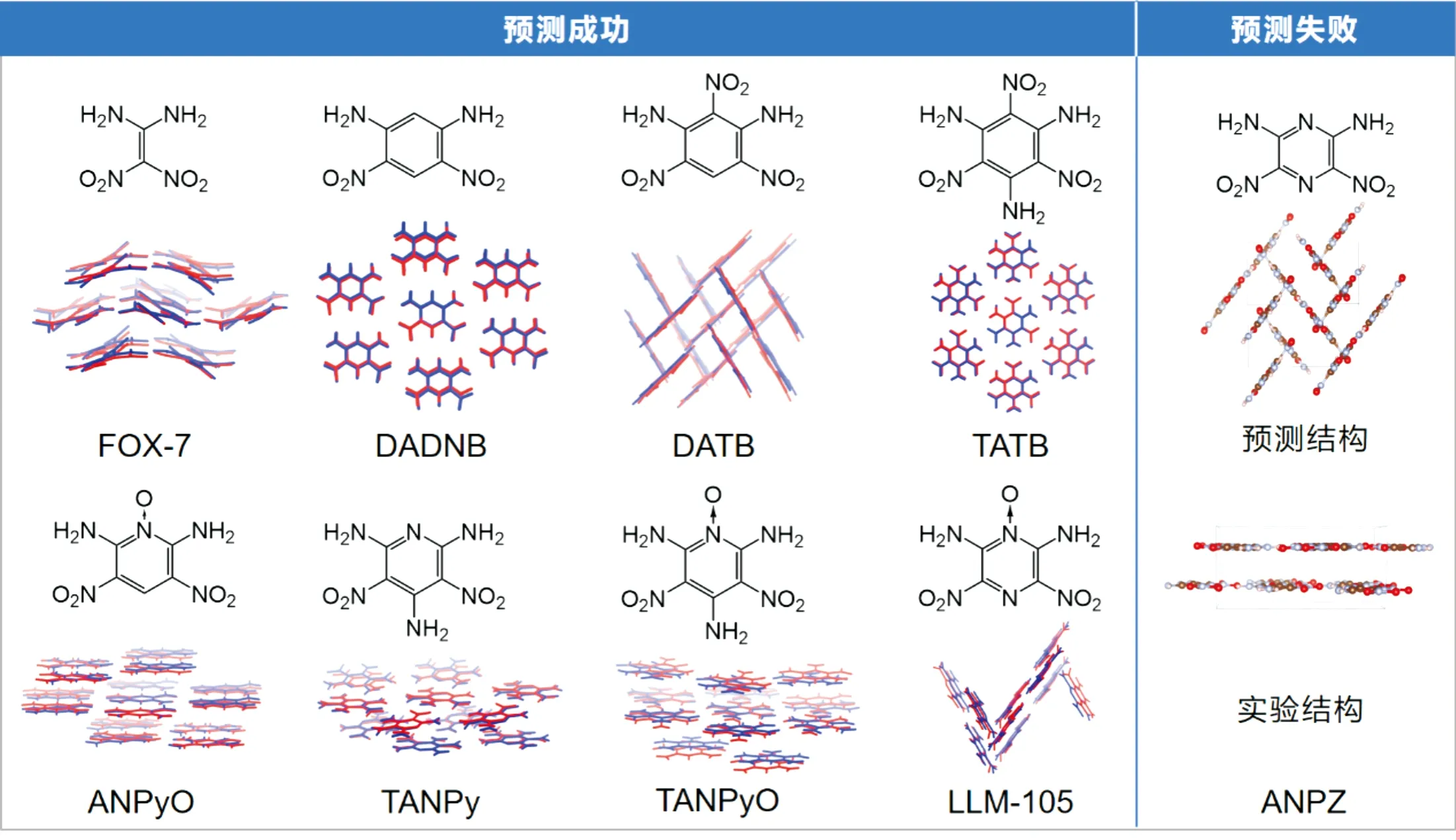
图7 九种典型硝基平面分子晶体结构及预测结构的叠加图,红色为实验结构、蓝色为预测结构[49]
1.3 机器学习设计策略
近年来,随着计算机硬件和大数据科学的高速发展,机器学习有望成为一种变革材料领域传统“试错”研发模式的新兴方法。利用机器学习可以从数据中自动地归纳规则或总结规律,从而建立材料影响因素与目标性质之间的映射关系,实现性能的预测与新材料的发现,并辅助科学家从不同的维度深入认知材料的机理特征。
在含能材料领域,机器学习研究的最新热潮可认为兴起于2018年,美国马里兰大学Elton等[62]建立了含能材料典型性质的多种预测模型,证明了机器学习从小样本数据集中获取含能材料信息的能力。2021年,中物院化材所Zhang的团队[68]利用机器学习实现了新型熔铸炸药载体材料的快速发现,通过随机森林等算法构建了5种含能材料性质的预测模型,从近4 000个候选分子中快速筛选出136个目标分子,并成功合成出其中8种,为新型含能材料的快速研发提供了一种新范式。
当前,机器学习主要用于含能材料结构与性质关联模型的构建[70-72],以实现目标性质的快速预估,包括晶体密度[62-69]、生成焓[62,63,66]、爆轰性能[6,62,63,67,68,73-76]、热分解温度[6,68]、撞击感度[77]等。其中,晶体密度是决定含能材料爆轰性能的关键参数,但鉴于准确预测晶体结构比较困难,通常采用基于量化计算的分子体积结合静电势修正的方法(DFT-QSPR)进行预估[69],平均绝对偏差约为0.044 g/cm3。因晶体密度具有相对较多的实验数据,受到了机器学习研究的较大关注。
如表2所示,研究人员通过不同的特征变量与算法构建了多种晶体密度的预测模型,计算精度可以达到与DFT-QSPR方法相当的程度,甚至更优。更为重要的是,与DFT-QSPR方法的耗时相比,机器学习预测模型的计算代价十分低廉,非常适用于高通量的虚拟筛选,从而加速新型含能材料的发现。例如,新加坡南洋理工大学等[66]利用建立的密度与生成焓机器学习模型,从含有约1.5亿个分子的PubChem数据库筛选出56个高能分子。加拿大麦吉尔大学Hong Guo等[78]通过建立爆热的机器学习模型,从PubChem和ICSD数据库中筛选出29个能量水平约为1.8倍TNT的候选分子。
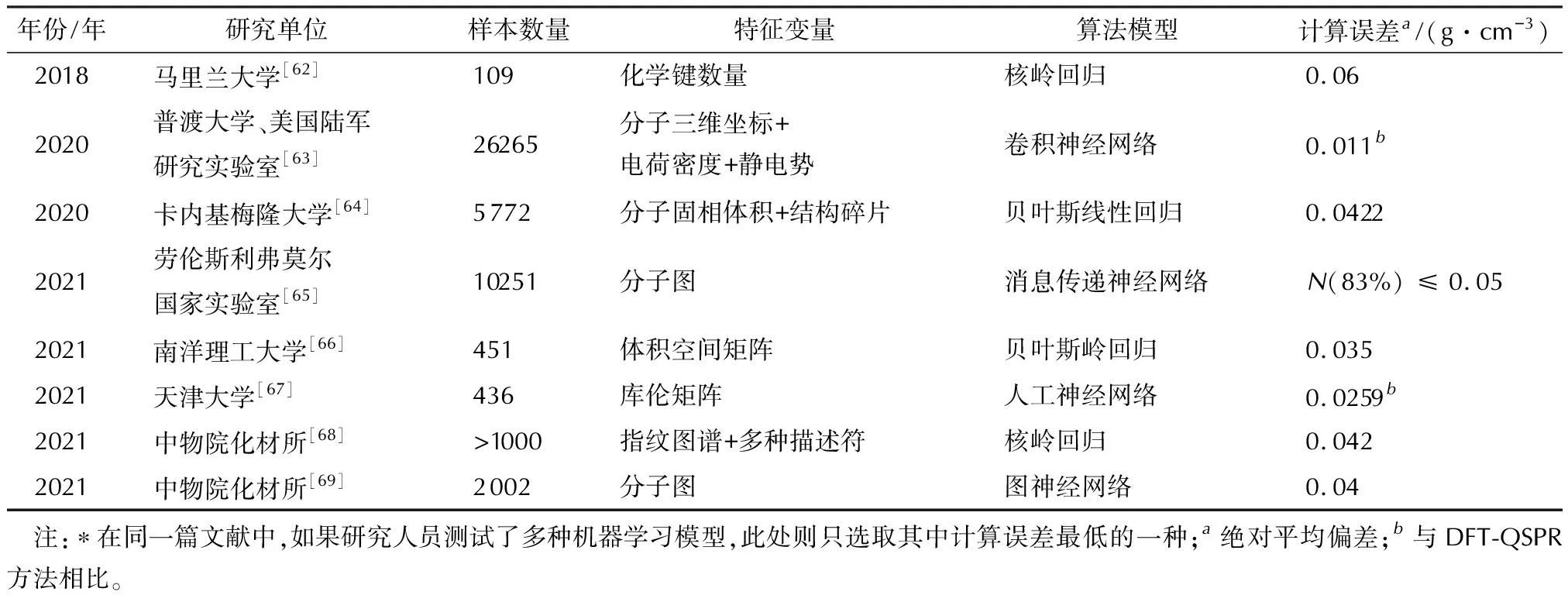
表2 机器学习在含能材料晶体密度预测中的典型应用*
值得注意的是,这些晶体密度预测模型中的特征变量仅与分子结构信息相关,说明机器学习算法可以在不知道晶体堆积结构的情况下较为准确地预测晶体密度。最近,四川大学Pu等[80]发展了一种新的图神经网络架构,可以仅基于两个分子的结构拓扑信息,预测二者能否形成共晶。通过对含能共晶、药物共晶、π-π共晶的独立测试,结果显示模型的预测准确率高达96%,并实验合成出一种新的CL-20基含能共晶。这表明机器学习能够基于实验已有数据自动建立分子结构与晶体堆积之间的关联,并快速准确地判断不同分子之间能否形成有效堆积结构。
在其他领域,基于图神经网络的机器学习被用于预测多环芳烃化合物的堆积模式,其模型准确率为76%,测试准确率为64%[81],为层状晶体的快速识别与筛选提供了一种新思路。2022年2月,中物院化材所Zhang的团队[79]在Engineering期刊上在线报道了机器学习指导合成层状含能材料的最近进展。该团队首先利用核岭回归算法构建了含能分子与密度、爆速、爆压、热分解温度等性质的机器学习模型,从25 112个分子中筛选出99个理论爆速大于8 400 m/s和热分解温度大于280℃的目标分子。此外,基于22个层状堆积和365非层状堆积实验晶体数据,借助SMILES生成的独热编码,利用卷积神经网络构建了晶体堆积模式的打分函数,用于预测形成“类石墨”堆积模式的概率,并进一步锁定了5个合成目标物(晶体堆积模式的打分均在0.5以上)。最终,成功指导实验合成出ICM-104。其晶体结构上具有显著的层状堆积特点,展示出了优异的综合性能(见图8)。由上述成功案例可见,利用机器学习进行层状含能材料的理性设计是一种极具潜力的策略。
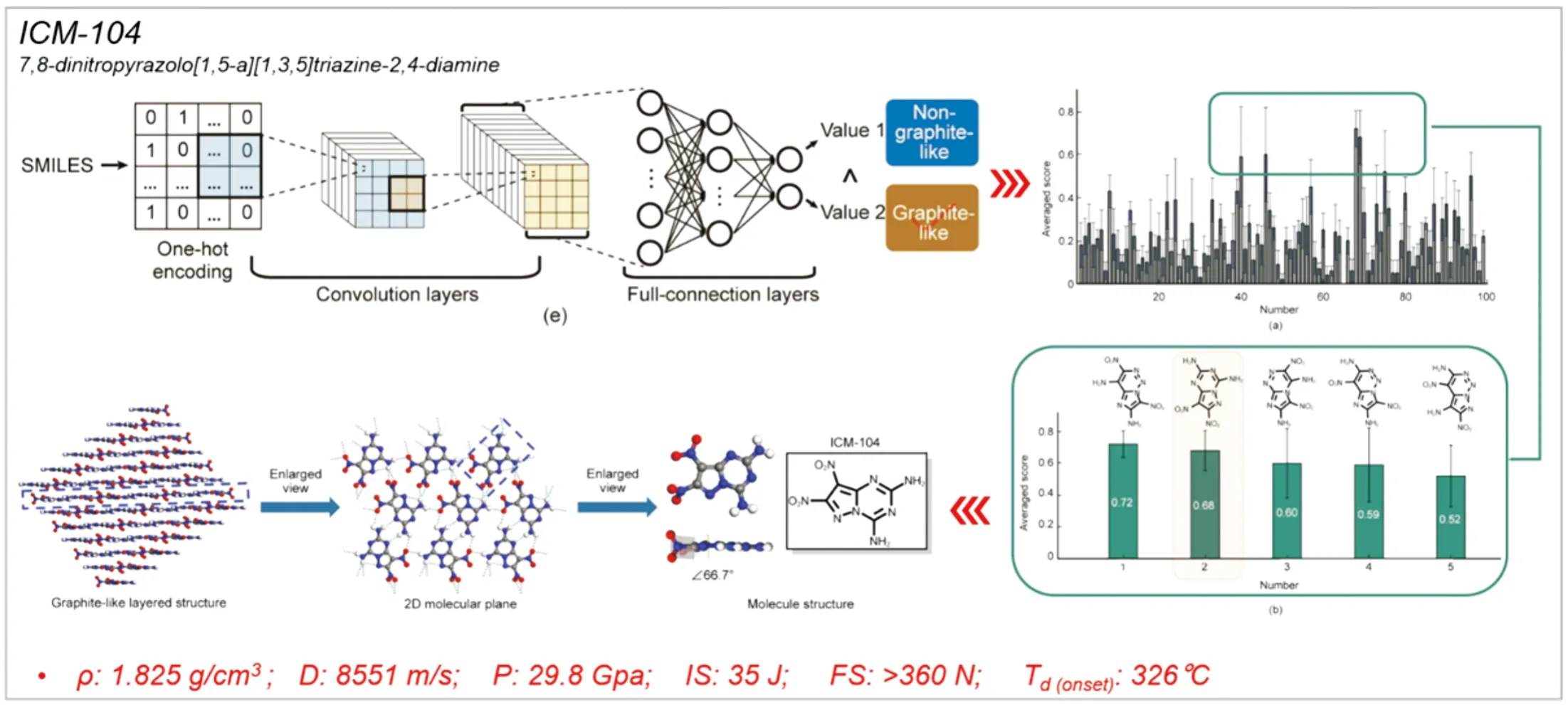
图8 机器学习指导合成“类石墨”含能材料ICM-104[79]
2 发展建议
当前,新型层状含能材料正快速发展,但其中大部分结构为离子盐或溶剂化物的多组分晶体,类似TATB的单分子层状含能晶体的构建仍然是重要挑战。为此,可从数据驱动的角度出发,深入理解层状晶体的堆积规律,发展基于机器学习的高通量设计方法。
2.1 基于数据库筛选深入理解构效关系
为实现从百万量级的晶体数据库中快速识别层状晶体结构,基于层状晶体结构的特点,建议从分子结构和晶体堆积两方面,从有机晶体数据库中对平面分子进行筛选,筛选判据可以从元素组成、物理性质以及构建的平面分子定量化表征和层状晶体结构定量化表征几个方面考虑。例如,本课题组通过如图9(a)所示的筛选流程,从CCDC数据库中的约106个晶体中,筛选出2 000个左右的晶体,筛选条件如下:
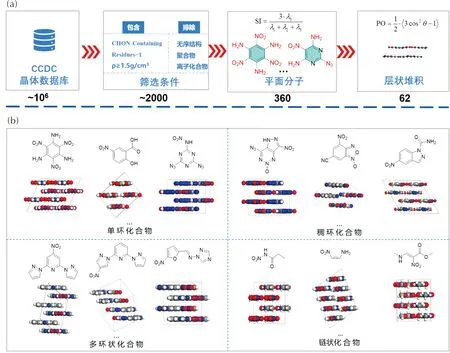
图9 (a)层状含能材料的筛选流程;(b)部分筛选得到的层状含能晶体
(1)包含CHON元素;
(2)是单质晶体,去除无序结构、聚合物和离子化合物;
(3)密度ρ≥1.5 g/cm3。
然后,从满足以上3个筛选条件的2 000个左右数据中,筛选得到形状指数(SI)≤0.01的360个具有平面分子形状的晶体,最后通过堆积取向系数PO(接近1)筛选得到62个层状堆积的晶体,部分结果如图9(b)所示。通过数据库的高通量筛选,有助于深入理解层状堆积含能材料的构效关系,也可为含能材料的机器学习设计策略提供足够的数据驱动力。
2.2 发展机器学习方法实现理性设计
随着各类机器学习和深度学习算法的不断开发,研究人员一方面可以尝试利用先进的算法训练获得较高精度的模型,另一方面可以通过提取特征分子描述符进一步提高预测精度。在层状含能晶体的预测模型中,主要为常见的特征描述符,例如氢键的供体和受体数目、π键等。最近,本课题组[82]通过研究10种分子结构相似的CHONF类含能化合物,提取出导致晶体堆积模式差异的主要因素O…C和O…N分子间相互作用。通过识别其在分子结构的结合位点,结合静电势表面分析发现,该位置为π轨道缺电子区域,即π-hole(如图10所示)。邻近的富电子NO2基团可形成π-hole分子间相互作用,因该种作用具有独特的方向性,导致了非层状晶体结构的形成。因此,未来可尝试将π-hole作为关键特征描述符用于模型训练。此外,探索挖掘更多的含能分子特征描述符可作为重要的研究方向之一。
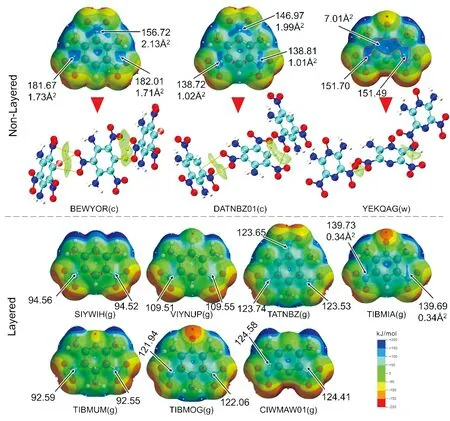
图10 10种CHONF平面分子的表面静电势图,箭头所指为π-hole区域的极大值
除特征描述符外,数据增强也是实现机器学习高效预测的重要因素。目前,层状含能材料的数量十分有限,因此,可以考虑引入合理的分子生成手段。本课题组建立了基于碎片组合的含能材料高通量分子生成方法,通过制定骨架与基团的键联规则,利用SMILES编码实现碎片结构之间的快速枚举组合,从而生成大量新型含能分子[30,83-85]。例如,本课题组曾以噁二唑等5个骨架、氧化偶氮桥连键、氟代偕二硝基等11个基团为碎片,通过限定骨架连接个数等规则,以单键键联的方式将不同碎片之间快速重组,共计生成了超过12万个多环CHONF含能分子[83]。这些高通量的分子生成策略可扩充层状含能材料的数据集,有望实现较好的机器学习训练效果。
3 结束语
本文从经验直觉、理论计算、机器学习3个方面综述了当前层状含能材料的主要设计策略,为后续层状含能材料设计理论与方法的丰富完善提供了参考,有助于加速新型层状含能材料的创制。更为重要的是,通过对层状含能材料的理论设计研究,将发展出晶体结构预测等新方法新技术,促进推动含能材料设计水平从分子层面提升至晶体层面,从而实现面向晶体尺度的含能材料理性设计。建议未来发展方向和研究重点如下:
(1)建立层状含能材料特征的定量化计算方法,实现数据库的高通量筛选,基于统计规律,深入理解构效关系;
(2)构建适合层状含能材料的特征描述体系,提升机器学习模型预测精度,实现高通量设计与筛选。
猜你喜欢
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
重型机械(2019年3期)2019-08-27
衡阳师范学院学报(2016年3期)2016-07-10
焊接(2016年9期)2016-02-27
新疆钢铁(2015年2期)2015-11-07
应用化工(2014年1期)2014-08-16
应用化工(2014年7期)2014-08-09
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年3期)2014-03-20
