
喷昔洛韦乳膏体外释放一致性研究
2023-06-07 06:39信长颖赵龙山多凯于新颖寻延滨刘利群黑龙江省药品检验研究院哈尔滨50088沈阳药科大学沈阳006
中南药学 2023年5期
信长颖,赵龙山,多凯,于新颖,寻延滨,刘利群*(. 黑龙江省药品检验研究院,哈尔滨 50088;.沈阳药科大学,沈阳 006)
喷昔洛韦是抗病毒药物,对水痘、带状疱疹病毒有很强的抑制作用[1]。喷昔洛韦乳膏剂属于半固体制剂,发挥治疗作用主要取决于递药的3个过程[2]:① 活性成分从基质中释放到第一层生物屏障-角质层,即体外释放(in vitro release,IVR);② 渗透扩散于角质层或其他皮肤层,即体外渗透(in vitro permeation test,IVP);③ 在作用部位产生期望的药理作用。2021年3月我国国家药品监督管理局药品审评中心发布了《皮肤外用化学仿制药研究技术指导原则(试行)》[3],该指导原则明确指出在对皮肤外用仿制药一致性评价时,应对关键质量属性(CQAs)体外释放试验(IVRT)和体外渗透试验(IVPT)进行研究。国外对阿昔洛韦乳膏的IVRT进行了详尽的研究[4-5],但国内多集中于在制剂研发初期采用IVRT和IVPT进行处方筛选[6-9]及综述[10],仍未见关于半固体制剂IVRT全面验证及等效性评价的实例分析。因此本文以2021年国家抽检的7个厂家的喷昔洛韦乳膏为例,采用Franz扩散池及与喷昔洛韦具有惰性的商品膜,研究该乳膏剂的体外释放,建立科学完善的方法学验证过程,评价国内市售喷昔洛韦乳膏与参比制剂体外释放速率的差异,为喷昔洛韦乳膏等半固体制剂开展质量一致性评价奠定基础。
1 材料
1.1 仪器
XS205DU型电子天平(十万分之一,梅特勒公司);精控型体外经皮渗透试验系统KX-V/HDP(大连科翔科技开发有限公司,带有12个竖式扩散池);UltiMate 3000高效液相色谱仪(赛默飞世尔公司)。混合纤维素微孔滤膜(0.45 μm,江苏绿盟科学仪器有限公司);尼龙微孔滤膜(0.45 μm,江苏绿盟科学仪器有限公司);Strat-M膜(直径25 mm,厚度300 μm,编号:SKBM02560,默克密理博)。
1.2 试验
对照品喷昔洛韦(批号:100769-201502,纯度:99.8%,中国食品药品检定研究院);甲醇(色谱纯,美国霍尼韦尔公司);磷酸二氢钾(分析纯,国药集团化学试剂有限公司);生理盐水(兰西哈三联制药有限公司)。
喷昔洛韦乳膏原研制剂(美国Perrigo,批号:I9210411,规格:10 g/支),作为参比制剂(R);2021年国家评价性抽检中涉及到的7个生产企业各1批样品作为仿制制剂(T1、T2、T3、T4、T5、T6、T7)。
2 方法与结果
2.1 接收液的选择
将过量的喷昔洛韦对照品溶解在生理盐水中,制备喷昔洛韦饱和溶液;将配制好的喷昔洛韦饱和溶液在(32.0±0.5)℃下恒温振荡24 h,实验平行3组,然后将饱和溶液取出离心,并用恒温至(32.0±0.5)℃的混合纤维素微孔滤膜过滤,将过滤好的溶液用生理盐水稀释并用HPLC分析,计算喷昔洛韦的饱和溶解度。结果喷昔洛韦在32℃生理盐水中的饱和浓度为1.975(SD:0.05)mg·mL-1,接收池体积为10 mL,接收液理论上可溶解约20 mg的喷昔洛韦;当喷昔洛韦乳膏的给药量约为0.2 g时,喷昔洛韦乳膏的规格为1%,样品中喷昔洛韦的给药量约为2 mg,用10 mL的生理盐水作为接收液进行体外释放试验可满足漏槽条件。
2.2 膜的选择
2.2.1 膜的惰性研究 在(32±0.5)℃条件下,分别将纤维素微孔滤膜、尼龙微孔滤膜、Strat-M膜浸入10 mL约为0.2 mg·mL-1的喷昔洛韦生理盐水溶液(上样0.2 g时,若喷昔洛韦全部释放,接收液中喷昔洛韦质量浓度为0.2 mg·mL-1)中保温24 h,平行3组。作为对照,制备没有膜的相同浓度的喷昔洛韦生理盐水溶液,同时保温24 h,平行3组。随后用HPLC测定各测试溶液的浓度,通过将放有浸渍膜的溶液的平均浓度除以对照品溶液的平均浓度,计算相对于对照的回收率,回收率应在(100.0±5.0)%,说明膜对喷昔洛韦无吸收[5]。结果混合纤维素微孔滤膜回收率为(98.5±0.9)%,尼龙微孔滤膜的回收率为(97.4±0.5)%,Strat-M膜的回收率为(97.8±0.8)%,3种膜对喷昔洛韦均无明显吸收。
2.2.2 膜的阻滞性研究 分别将混合纤维素微孔滤膜、尼龙微孔滤膜、Strat-M膜(各膜均预先在接收液中浸泡30 min以消除气泡)固定于Franz扩散池的一端,接收池接收液总体积为10 mL,扩散池上精密加入0.2 mg·mL-1的喷昔洛韦生理盐水溶液0.2 mL,在600 r·min-1和(32±0.5)℃条件下恒速恒温搅拌,并于0.5、1、2 h取接收液0.2 mL,每次取样完成后立即补加0.2 mL(32±0.5)℃的接收液,根据测定每一取样点喷昔洛韦的浓度,计算药物溶液累积释放率。以累积释放百分率对时间t作图,结果见图1,喷昔洛韦溶液在混合纤维素微孔滤膜上的释放速率最大,无阻滞作用,因此选择混合纤维素微孔滤膜进行研究。
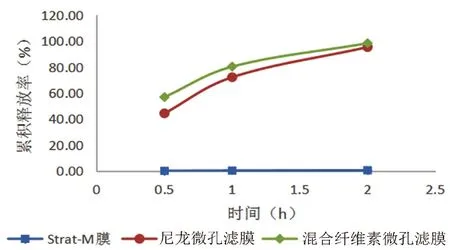
图1 昔洛韦溶液在不同商品膜上的释放情况Fig 1 Release of penciclovir solution on different commercial films
2.3 体外释放的测定方法
2.3.1 方法 采用Franz扩散池[11](《美国药典》USP43中的C型立式扩散池)进行体外释放试验,装置示意图见图2。将混合纤维素微孔滤膜(预先在接收液中浸泡30 min以消除气泡)固定于扩散池的一端,精密称取约0.2 g喷昔洛韦乳膏,在释药面积1.77 cm2的膜上通过定量环均匀涂抹上样,接收池接收液总体积为10 mL,在600 r·min-1和(32±0.5)℃条件下恒速恒温搅拌,并于0.5、1、2、4、6 h取接收液0.2 mL[每次取样完成后立即补加0.2 mL(32±0.5)℃的接收液],分别作为体外释放测定用供试品溶液,采用HPLC法进行定量分析。每个样品运行6次,每次在不同的扩散池上运行,并与参比制剂进行对比,以消除不同扩散池带来的差异。
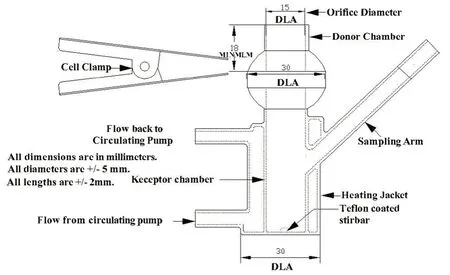
图2 C 型立式扩散池[11]Fig 2 Vertical diffusion cell model C[11]
2.3.2 方法学验证[14]按FDA的阿昔洛韦乳膏指南草案方法学验证方法对本IVRT方法进行方法学验证。
①日内精密度和日间精密度:精密称取参比制剂6份,分别置于6个扩散池,计算每个扩散池的释放速率,6个释放速率的变异系数(CV)应小于15%,即为日内精密度。次日,再次精密称取参比制剂6份,分别置于6个扩散池,计算每个扩散池的释放速率,计算3 d共18个释放速率的CV,即为日间精密度。结果日内精密度的CV为4.6%,日间精密度的CV为9.2%,均小于15%。符合检测重现性的要求。
② 线性:“①”项下18个释放曲线方程,每个方程的r2均大于0.99,满足指南草案中要求的r2大于0.90的要求,说明本IVRT方法可采用Higuchi方程进行线性拟合计算。
③ 灵敏性:按某一已知企业的喷昔洛韦乳膏的制备方法制备0.5%、1.0%、1.5% 3种不同规格的喷昔洛韦乳膏,每个浓度测定6份,计算平均释放速率,0.5%乳膏的释放速率为143.8 μg·cm-2·h1/2,1%乳膏的释放速率为241.3 μg·cm-2·h1/2,1.5%乳膏的释放速率为352.1 μg·cm-2·h1/2。说明该方法是灵敏的。
④ 特异性:以“①”项下0.5%、1.0%、1.5%制剂的释放速率为纵坐标,以标示百分含量为横坐标,线性回归方程为Y=2.07×104X+35.87,r2为0.9989。可见本方法具有特异性,能准确监测制剂中释放速率随药物浓度变化的比例关系。
⑤ 选择性:取“①”项下喷昔洛韦乳膏,分别计算0.5%、1.0%、1.5%与1.0%乳膏的90%置信区间,1.0%乳膏与1.0%乳膏相比,90%置信区间均在75.00%~133.33%内,但新制备的0.5%、1.5%的喷昔洛韦乳膏的释放速率与1.0%乳膏相比,在75.00%~133.33%外,可见本方法具备足够的选择能力,可以识别处方中主药浓度的变化。
⑥ 耐用性:测定温度为(34±0.5)℃、(30±0.5)℃时,转速为540、660 r·min-1时的释放速率,与原条件相比,平均释放速率的CV分别为10.2%、11.5%、12.0%、14.0%,均小于15%,说明本方法的耐用性良好。
2.4 高效液相色谱法测定喷昔洛韦的含量[12]
2.4.1 方法 采用十八烷基硅烷键合硅胶为填充剂,流动相为0.02 mol·L-1磷酸二氢钾溶液-甲醇(9∶1);流速1.0 mL·min-1;检测波长254 nm;柱温30℃;进样量10 μL。
2.4.2 方法学验证
① 专属性:精密称取喷昔洛韦对照品12.91 mg置100 mL量瓶中,用生理盐水超声使溶解并稀释至刻度,摇匀,作为对照品溶液。精密量取“2.3.1”项下的供试品溶液、对照品溶液及空白溶剂10 μL进样分析,典型色谱图见图3。空白溶剂对测定无干扰,本方法专属性好。
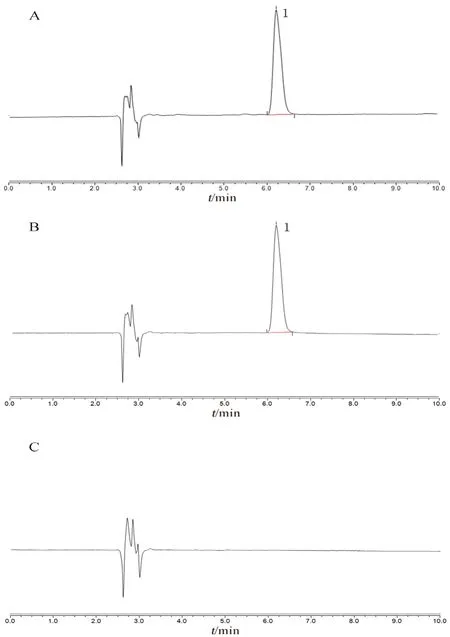
图3 喷昔洛韦的高效液相色谱图Fig 3 HPLC chromatograms of penciclovir
② 线性:精密称取喷昔洛韦对照品12.91 mg置100 mL量瓶中,用生理盐水超声使溶解并定容至刻度,摇匀,作为喷昔洛韦对照品储备液。精密量取喷昔洛韦对照品储备液0.1、1、5、7.5、10 mL置10 mL量瓶中,用生理盐水稀释至刻度,摇匀,即得标准系列溶液,精密量取各溶液10 μL进样分析,绘制标准曲线。以质量浓度(μg·mL-1)为横坐标(X),以峰面积为纵坐标(Y),线性回归方程为Y=505.7X-0.001 81(r=1.000),喷昔洛韦在0.0013~0.1288 mg·mL-1与峰面积呈良好的线性关系。
③ 精密度:分别精密量取对照品溶液10 μL,连续进样5次,5次峰面积RSD为0.20%,符合测定要求。
④ 准确度:取6份参比制剂R约0.2 g,置于20 mL量瓶中,各分别精密加入约2 mg喷昔洛韦对照品,超声使溶解,用生理盐水稀释至刻度。精密量取各溶液10 μL进样分析,结果得回收率平均值为95.5%,RSD为3.5%,满足测定要求。
⑤ 重复性:取6份参比制剂R,置于10 mL量瓶中,用生理盐水溶解并稀释刻度,摇匀,精密量取各溶液10 μL进样分析,结果得峰面积RSD为3.3%。可认为本方法重复性好。
⑥ 稳定性:取“⑤”重复性项下供试品溶液分别放置0、4、8、12、24 h测定,结果得峰面积的RSD为0.50%,稳定性符合测定要求。
⑦ 检测限和定量限:精密量取专属性项下的对照品溶液,反复稀释,以喷昔洛韦色谱峰信噪比为3∶1时为检测限,检测限为0.32 ng,以信噪比为10∶1时为定量限,定量限为0.64 ng。
2.5 体外释放速率和累积释放率的计算及统计分析方法[13]
实验过程中每次取样测得的接收介质中的药物浓度,按以下公式计算单位面积的累积释放量:
Qn=每单位面积在时间(n)释放的量(μg·cm-2);Cn=不同采样时间(n)下接收介质中的药物浓度(μg·cm-3);Vs=样品体积(cm3);Vc=接收室容积(cm3);Ac=有效扩散面积(cm2)。
半固体制剂中的药物释放一般遵循Higuchi方程,释放达稳态后单位面积累积释放量与时间的平方根呈线性关系,其相关系数r2应大于0.90,直线部分的斜率为药物释放速率。
将各取样点的累积释放量,除以加入扩散池中的药物的实际量,计算各时间点的累积释放率。体外释放参比制剂(R)和仿制制剂(T)各产生6个斜率(体外释放速率)。计算仿制制剂的体外释放速率与参比制剂的体外释放速率比值的90%置信区间,用百分比表示。基于SUPAC-SS指南中描述的Mann-Whitney U检验,来判断两组数据的中位数有无差别。Mann-Whitney U检验是将两组样本的数值统一由小到大编秩,由Mann-Whitney U检验求出90%置信区间的上下界值的范围。如上下限均落在75.00%~133.33%,则表示仿制制剂与参比制剂的释放速率的中位数差异在允许范围内,仿制制剂与参比制剂的释放速率是等效的,如置信区间的值仍落在75.00%~133.33%外,则判断为两制剂的释放速率不等值。
2.6 仿制制剂与参比制剂的释放结果比较
以不同时间累积释放量(μg ·cm-2)为纵坐标Y,以时间的平方根为横坐标X,对其进行线性回归,参比制剂与7个仿制制剂的释放速率方程见表1,方程的斜率为释放速率。各仿制制剂与参比制剂释放速率的比较见图4。7家仿制制剂与参比制剂(T/R)释放速率比值的90%置信区间上下限及一致性判定见表2。可知,7家仿制制剂中,仅一家企业的制剂(T4)与参比制剂的释放速率一致,其他6家仿制制剂均不一致。其中T1、T2、T5、T6、T7释放速率高于R,而T3释放速率低于R。

表1 释放速率方程Tab 1 Release rate equation
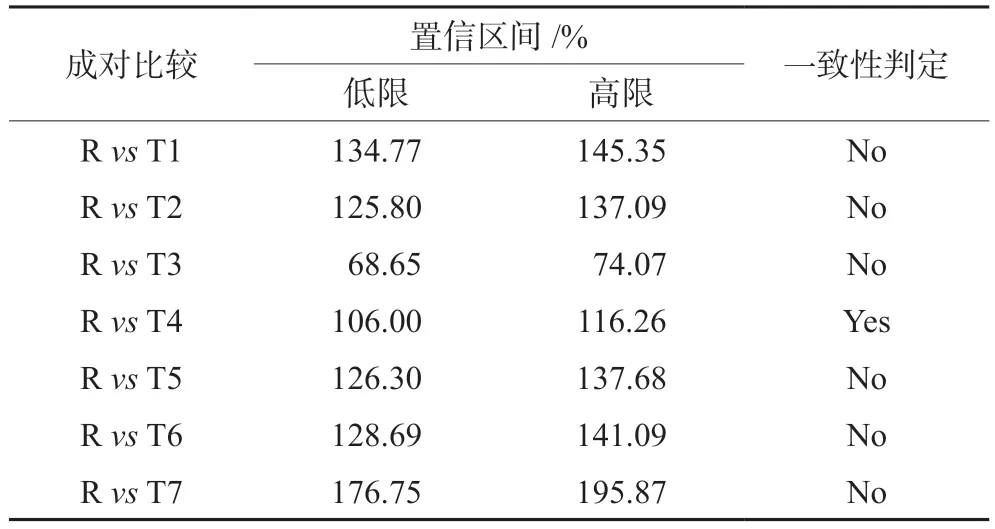
表2 7家仿制制剂与参比制剂释放速率比值的90%置信区间及一致性判定Tab 2 Confidence interval and consistency of release rate of generic preparation from 7 manufacturers and reference preparation
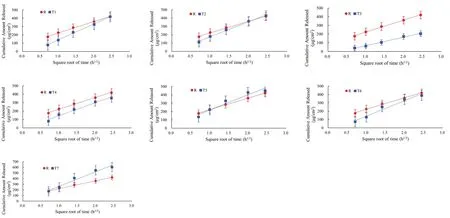
图4 7家仿制制剂与参比制剂释放速率的比较Fig 4 Release rate of generic preparation from 7 manufacturers and reference preparation
3 讨论
FDA 在其颁布的SUPAC-SS 指导原则“非无菌半固体制剂扩大规模和上市后变更:体外释放试验和体内生物等效性要求”中指出,半固体局部用制剂在生产放大和产品批生产发生变更时须进行体外释放试验,以保证产品质量的一致性和等效性[13]。FDA已发布了多个外用半固体仿制药的指南草案,如阿昔洛韦乳膏、磷酸克林霉素凝胶、甲硝唑阴道用凝胶、维甲酸凝胶、贝沙罗汀凝胶和二十二烷醇乳膏等,完成IVRT或IVRT和体外渗透试验后可申请生物等效性豁免[14-19]。
本文以喷昔洛韦乳膏作为模型药物,建立了乳膏体外释放测定方法,在方法建立之初,考察了20%丙二醇的生理盐水溶液、5%吐温80溶液以及生理盐水3种接收液,发现因3种接收液均达到漏槽条件,参比制剂在3种接收液释放速率无差异,从一定程度说明IVRT作为半固体制剂的一种物理性质,接收介质不影响制剂的形态,即接收介质不影响药物的释放,合适的接收介质可以是多种,在选择接收介质时,只要保证溶解度要求及接收介质的稳定性即可。
IVRT试验选择膜时主要取决于三点:膜的惰性(与API和接收液无反应)、无扩散、无阻滞。因此本文设计了膜惰性试验,所选择的3种膜与喷昔洛韦均没有结合情况。但在膜的阻滞性研究中,3种膜在2 h后均对喷昔洛韦无阻滞,但在纤维素微孔滤膜上速度快,阻滞作用小,因此本文选择纤维素微孔滤膜进行IVRT研究。喷昔洛韦溶液2 h在Strat-M膜中基本无透过,此外本次还监测了参比制剂与各仿制制剂在Strat-M膜24 h的释放情况,参比制剂和各仿制制剂在该膜上的释放量均低于检测限,Strat-M膜与人体皮肤相似,适用于IVPT研究[20],不适用于药物释放的考察,同时证明各制剂均不透过皮肤,与说明书标注“本品在血液中无法检测到”一致,证明了各制剂的安全性[21]。
体外释放速率可能不能完全反映药物透皮递送的速率,但可参考检测到的释放速率的变化来推测某些物理化学性质(如原料药粒度、物理状态)及工艺参数等变化带来的制剂内在质量差异,此类差异可能会最终影响制剂的疗效。并且有国外学者提出,如果仿制药与参比制剂辅料的种类(Q1)、用量(Q2)不同,即使辅料存在差异,但辅料是惰性的,对药物的透皮没有影响,如果反映物理性能与微观结构(Q3)的体外释放率相同,也可以申请生物等效豁免,这一理论是由提出生物药剂学分类系统(BCS)理论的研究小组成员提出,在FDA网站可以查到多次关于这一分类系统局部给药分类系统(TCS)的研讨,可见体外释放试验对于半固体制剂研究的重要性[22]。
本文首次建立了IVRT的方法学验证,采用伪无限上样量(取样最后时间点的累积释放率小于30%),不同浓度的喷昔洛韦乳膏释放速率不同,验证了方法的灵敏性,释放速率与浓度的变化呈线性关系验证了方法的特异性,该方法可区分不同浓度的喷昔洛韦乳膏,说明建立的方法具有选择性,进一步证明了所选用的膜惰性无阻滞。此外本试验还控制环境温度(21±2)℃,湿度(50±20)%,并在试验前对扩散池的体积、孔口直径、转速、工作台的倾斜度进行了全面的验证。经验证的IVRT方法可准确评价产品的质量,但同时建议采用氢化可的松校正软膏对扩散池仪器进行更为全面的验证(类似于溶出度中的水杨酸校正片)[23]。
4 结论
本研究采用IVRT方法测定了喷昔洛韦乳膏国内7家仿制企业的各1批样品与参比制剂的释放速率,并按照FDA的SUPAC-SS指导原则中的等效性判定方法对各试验制剂与参比制剂的等效性进行判定,结果仅1家企业制剂的释放速率与参比制剂在等效性范围75.00%~133.33%内,其他6家企业制剂的释放速率与参比制剂均不等效。虽然在IVRT研究中测定的特定释放速率并不与药物在体内从产品中释放的速率相关,但经过验证的IVRT方法可以对试验制剂和参比制剂之间的释放速率差异敏感[24]。如果在IVRT研究中观察到这种差异,则增加了产品性能可能在体内存在的潜在差异。相反,与参比制剂相比,试验制剂具有相同的药物释放率,则降低了生物等效性失败模式的风险,因此支持生物等效性的论证。本研究为进一步扩大建立乳膏体外释放测定方法及质量一致性评价奠定基础。
猜你喜欢
皮肤病与性病(2021年3期)2021-07-30
皮肤病与性病(2021年3期)2021-07-30
陶瓷学报(2020年3期)2020-10-27
国外医药(抗生素分册)(2016年3期)2016-07-12
中国继续医学教育(2015年6期)2016-01-07
中国当代医药(2015年8期)2015-03-01
应用化工(2014年1期)2014-08-16
云南中医学院学报(2014年5期)2014-07-31
中国药业(2014年21期)2014-05-26
