
耳聋基因检测在新生儿听力筛查中的应用
2023-05-26 08:23栾海萍
医学理论与实践 2023年10期
栾海萍
上海市普陀区妇婴保健院检验科 200062
听力障碍是最常见的出生缺陷之一,国内外报道发生率为1‰~3‰,经过ICU抢救过的新生儿发生率更高。若是按照我国每年出生1 900万新生儿计算,每年新增2万~3万听力障碍儿童[1-4]。听力障碍后果不仅在于“聋”,而且在于“哑”,特别是迟发性耳聋发病率高,在新生儿中发病率为1.33‰,4岁儿童的发病率为2.7‰,青少年中为3.5‰。研究发现我国耳聋基因的热点谱存在地域差异性[5],本研究以近年来出生的2 916例新生儿为研究对象,采用PCR扩增法和导流杂交相结合的方法,对耳聋基因GJB2、GJB3、SLC26A4、mtDNA的13个位点进行检测。
1 对象与方法
1.1 研究对象 选择2018年6月—2022年6月在我院出生的2 916例新生儿为研究对象,其中男1 516例,女1 400例。新生儿家长均在新生儿科医生充分告知的情况下,自愿签署知情同意书。
1.2 研究方法
1.2.1 标本采集:由新生儿医生采集,采集新生儿脚底末梢血制成血斑卡,晾干后每张血斑卡使用独立的自封卡,送至检验科标本接收处,通过签收血斑卡保证检验前质量控制。
1.2.2 核酸提取:采用打孔器对血斑卡均匀打取3~4个干血片置于1.5ml离心管中,采用凯普生物全血细胞DNA提取试剂盒的离心柱法提取DNA。
1.2.3 核酸扩增及导流杂交:对13个GJB2(35delG、176-191del16、235delC、299-300delAT、155delTCTG),SLC26A4(2168A>G,IVS7-2A>G、1229C>T),GJB3(538C>T),mtDNA(1494C>T、1555A>G、12201T>G、7445A>G)基因位点设计引物,采用PCR扩增仪为Bioer Life Express进行扩增反应。并对扩增反应物通过变性,对变性产物用核酸分子杂交仪HB-2012A杂交反应,并设置相应质控点,保证杂交质量。
1.2.4 验证方法:为保证结果的准确性,杂合突变采用重新提取DNA复查的办法,纯合突变和异常突变(正常点和突变点均未显色)Sanger测序。使用聚合酶链式反应技术(PCR)对突变基因目标位置进行扩增,并采用特异性引物进行 Sanger 测序检测。特异性引物的设计是专门针对目标位点,并覆盖该位点上下游的序列;标本检测得到的序列与 NCBI 所提供 基因参考序列进行比对和注释。突变的表述参照 Human Genome Variation Society (HGVS) version 15.11进行。
1.2.5 质量控制:采用空白对照作为阴性质控﹑以往阳性标本作为阳性质控监测扩增过程,杂交过程由生物素标记点作为监测。并参加上海市临检中心室间质控作为实验室质量保证。
2 结果
2.1 筛查结果分析 2 916例新生儿共筛查出138例耳聋基因突变,总阳性率为4.7%,GJB2-235基因突变率最高。上海普陀地区常见的耳聋基因GJB2和SLC26A4,热点突变位点是GJB2的235delC、SLC26A4的IVS7-2A>G位点。其他复合突变:mtDNA-1555纯合突变、SLC26A4-IVS7-2杂合突变的复合突变。GJB2-235的杂合突变、SLC26A4-IVS7-2杂合突变的复合突变。见表1。
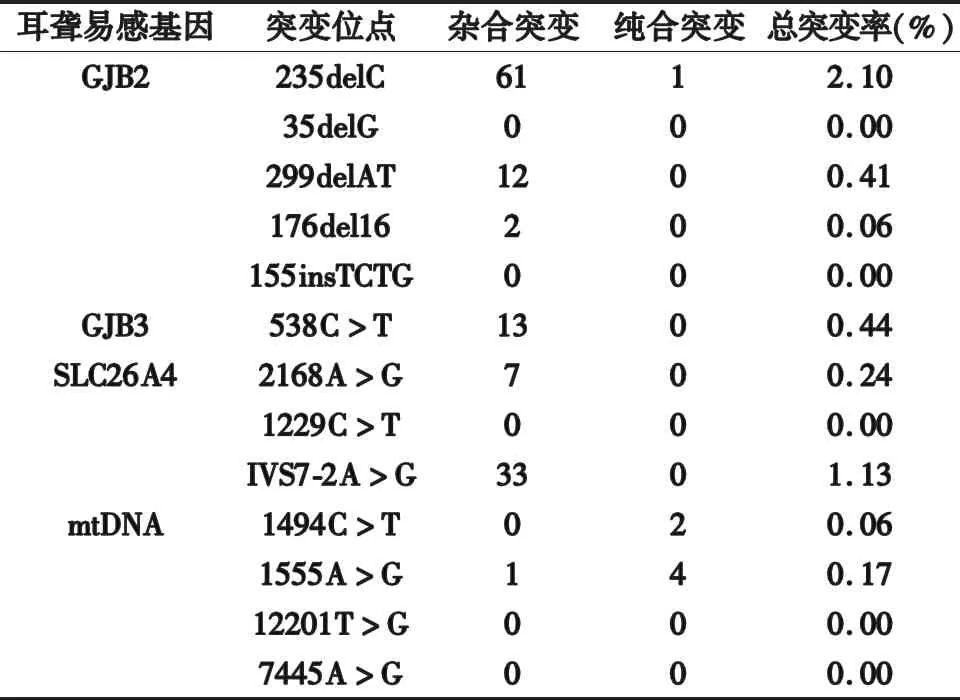
表1 2 916例新生儿耳聋基因筛查突变表
2.2 异常突变点一代测序验证结果 在2 916例标本中,有6例异常突变的标本,分别是1.7445正常点和杂合突变点均未显色,Sanger测序为7443均质突变,其突变影响了探针结合,该突变为无义突变,不致病。2.1494正常点和杂合点未出,Sanger测序为1503处发生均质突变。3.7445正常点显色很淡,Sanger测序为7443(A>G)的多态性。4.1494和1555正常点显色很淡,Sanger测序为mtDNA m.1709G>A和m.1716T>C均质突变,致病性不明。1494位点和1555位点未见突变。5.7445正常点和杂合突变点均未显色,Sanger测序为7443A>G,其突变影响了7445探针结合,该突变致病性不明,见图1。6.1494点淡,测序显示多态性。
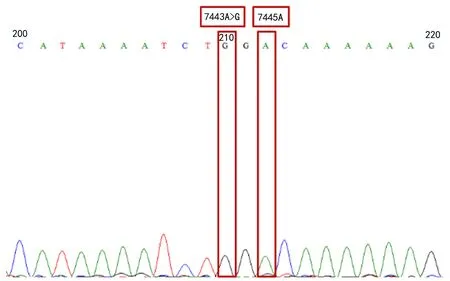
图1 mtDNA基因7445位点测序图
2.3 纯合突变一代测序验证结果 在2 916例标本中,有7例纯合突变,一代测序验证情况均符合杂交结果,其中1例GJB2-235纯合突变,通过随访发现该新生儿两耳听力未过。由于该突变一般无法通过物理听力筛查,听力基本丧失,经确诊后建议进行干预治疗(佩戴助听器,人工耳蜗植入术)。此外,做好婚育或产前遗传咨询,即将来其配偶也应进行婚育前耳聋基因检测,必要时可进行产前诊断以确认后代耳聋基因的携带情况。由于我院没有耳鼻喉科,故转诊到上级医院进行随访,后续随访结果为尽快植入人工耳蜗。其余6例听力筛查均通过,线粒体DNA均质突变患者应终生禁用耳毒性药物,见图2、3。

图2 导流杂交膜条

图3 耳聋基因mtDNA1555均质突变测序图
3 讨论
听力障碍作为残障性疾病,在新生儿群体中较常见。新生儿听力筛查作为新生儿重点筛查疾病之一,卫生部于2010年颁布的《新生儿听力筛查技术规范》中提出,应尽早发现听力障碍个体,予以及时、有效干预,以防聋致哑[6]。近年来,随着新生儿听力筛查的研究不断深入,发现单纯听力筛查可
能造成药物性耳聋基因携带者以及迟发性耳聋患儿的漏诊,因此,自2007年临床推荐在常规听力筛查基础上应联合基因筛查。遗传性耳聋分为综合征性耳聋和非综合征性耳聋,其中非综合征性耳聋约占70%。根据流行病学调查,非综合征性耳聋绝大多数为单基因遗传病,我国非综合性耳聋主要由GJB2、GJB3、SLC26A4、mtDNA引起[7-9],所以针对新生儿的耳聋基因和听力筛查可以了解他们是否携带耳聋基因和听力损失情况。通过进一步检测来决定是否需要干预性治疗,让儿童不错过学习语言的最佳时期。由于听力下降是一个过程,所以对于纯合突变和复合杂合突变的病例,也应建议进一步进行耳鼻喉科的随访。耳聋严重影响个人的生活质量,降低生活幸福指数。
在2 916例耳聋基因筛查中,总阳性率为4.7% ,阳性位点携带率与国内文献基本接近[10-11]。巫静帆等[12]对33 810例新生儿听力筛查与聋病易感基因筛查进行分析时发现,GJB-2基因突变发生率最高,为1.96%。另一项研究显示GJB-2基因突变率最高,突变频率为2.57%,GJB2纯合突变引起的先天性重度耳聋使用耳蜗干预性治疗,效果良好[13]。本研究中,GJB-2基因突变点最高,为2.1%,杂合突变点共61例,其结果与之相符。SLC26A4基因突变率为1.37%,该基因即“一巴掌致聋”,带有该基因的病人应避免外力碰撞对耳朵的伤害。研究表明,SLC26A4基因是仅次于GJB-2基因突变所致的感音神经性聋的遗传学病因[14]。SLC26A4基因突变与大潜艇导水管综合征存在密切关联,其典型表现为前庭导水管扩大,同时伴有感音神经性或混合性耳聋,其纯合突变、双杂合突变均是致聋的重要因素。GJB-3基因是国内本土克隆和鉴定的第一个遗传聋病基因,易引起迟发性聋病,多数患者在青年时期表现为听力正常,随着年龄增长进入青壮期听力逐渐降低,直至发展为重度聋病,其基因突变的原因与高频听力下降相关。李欣欣[15]研究结果显示,GJB-3基因突变为0.27%,该研究证实该基因为常染色体显性遗传,携带此基因新生儿应长期监测听力问题,以便尽早发现听力异常。本研究中,GJB-3基因突变率为0.44%,耳聋基因突变被证实与高频听力下降有关,一般发病为青少年期或者成人期,是一种迟发性耳聋基因。mtDNA基因属母系遗传,常见的两种突变类型为1494C和1555A,且存在同质性和异质性突变,两种突变本研究中突变率为0.23%,带有该突变基因的新生儿禁止使用氨基糖苷类药物。
一个耳聋患者会耗费大量的社会资源,大多数听力缺陷儿童来自听力正常家庭,由于听力筛查具有太多局限性,结合耳聋易感基因筛查可以提早干预,避免“一巴掌致聋”和“一针致聋”的悲剧发生。
猜你喜欢
中国民间疗法(2021年8期)2021-07-22
种子(2021年3期)2021-04-12
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年2期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
外语教学理论与实践(2016年1期)2016-06-11
中国中医药现代远程教育(2014年13期)2014-03-01
河南医学研究(2014年5期)2014-02-27
