
1例表现为肾病综合征的迟发型法布雷病
2023-05-26 08:13张鹏伟高佩娟马志刚
医学理论与实践 2023年10期
程 霞 张鹏伟 高佩娟 马志刚
1 甘肃中医药大学第一临床医学院,甘肃省兰州市 730000; 2 甘肃省人民医院肾病科
法布雷病(Fabry病)为一种罕见病,是由编码α-半乳糖苷酶的GLA基因突变引起的X染色体连锁性溶酶体贮积症,以神经酰胺三己糖苷为主无法正常代谢,渐进性的沉积于人体各组织及器官如皮肤、心脏、肾脏、神经系统等,从而使机体产生一系列渐进性损害。其发病率低且临床工作中极易误诊,本文报道了1例迟发型法布雷病并对该疾病进行文献复习。
1 病例资料
患者男性,33岁,因发现蛋白尿6年收住入院。自诉入院6年前体检时发现尿蛋白3+,偶伴少量泡沫尿,余无特殊不适。当时就诊于某市三甲医院查“白蛋白44g/L 尿素氮3.5μmol/L 血肌酐76.9μmol/L 24h尿蛋白4g(血脂及其他数据不详),肾穿刺活检示局灶硬化型”,诊断为肾病综合征予以强的松、代文(缬沙坦)、来氟米特等药物治疗(强的松起始剂量50mg,服用5~6个月后逐渐停药,时长为1年左右)。2015—2020年尿蛋白未见明显转阴,24h尿蛋白波动于0.5~3.5g之间。近1年尿蛋白较前加重,为行第2次肾组织活检术收住我科。既往行肾穿刺活检术、阑尾切除术。婚育史及家族史无特殊。入院查体:生命体征平稳,体格检查未见异常。
辅助检查:血清白蛋白39g/L,血清肌酐35.8μmol/L,胆固醇6.52mmol/L,甘油三酯2.04mmol/L,低密度胆固醇4.46mmol/L。24h尿蛋白定量:3.44g。SDS-尿蛋白电泳:尿中分子量蛋白88.7%,尿大分子量蛋白11.3%。血常规:血红蛋白181g/L,血小板340×109/L。免疫功能:IgG 7.15g/L,IgM 2.98g/L。抗核抗体谱、抗磷脂酶A2受体抗体、抗肾小球基底膜抗体、抗中性粒细胞胞浆抗体、血清蛋白电泳、血清免疫固定电泳等均未见异常。心电图示:窦性心律,电轴正常,异常心电图 ST-T改变。腹部及泌尿系超声:双肾形态、大小、位置如常,左肾大小105mm×55mm,右肾大小117mm×68mm。双肾实质回声略增强,皮髓质界限尚清,内未见肿物影,余未见异常。心脏彩超:左房、右室增大,左室向心性肥厚,二尖瓣关闭不全(轻度),三尖瓣返流(轻度),左室收缩功能正常[EF(Teich)=66%,FS=36%]。
肾组织活检:光镜:可见8个肾小球,其中1个肾小球球性硬化,2个肾小球节段性硬化,其余肾小球系膜细胞和基质轻度弥漫性增生,足细胞明显空泡变性。肾小管上皮细胞颗粒及空泡变性,个别肾小管基底膜增厚,管腔缩小,小灶状萎缩,肾间质小灶状炎症细胞浸润伴轻度纤维化,小动脉管壁增厚,管腔狭窄(见图1)。特殊染色:刚果红、氧化刚果红、光镜a:PAS,×200 b:PASM,×400 c:Masson,×400;足细胞明显空泡变性,呈泡沫状,肾小管上皮细胞颗粒及空泡变性红“O”均为阴性。免疫荧光:IgG(-)、IgA(-)、IgM(+)、C3(-)、C1q(-)、kappa(-)、lambda(-)、PLA2R(-)、TSHD7A(-)。电镜:毛细血管内皮细胞明显空泡变性。肾小囊壁层细胞空泡变性,无明显增生。肾小球基底膜节段性增厚,足突弥漫融合,部分足细胞空泡变性呈泡沫状,次级溶酶体增多并见大量髓样小体和斑马小体(见图2),未见电子致密物沉积。最后报告:高度疑为Fabry病肾病,建议临床完善相关基因检查。

图1 肾组活检
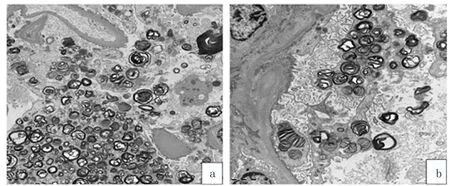
图2 肾组活检
溶酶体贮积症检验示:α-Gal A为0.41(参考范围2.4~17.65),结果提示α-Gal A低于正常值范围,符合Fabry病的改变。
基因检测:在GLA基因上检测出1个c.902G>A(p.Arg301Gln)的半合子变异位点,此变异为错义变异,在Clinvar数据库中被收录为致病变异,c.902G>A变异在男性中通常会导致迟发型Fabry病,患者在男性通常在成年期出现心律失常、左心室肥厚(LVH)或蛋白尿以及肾功能不全导致肾衰竭。
患者入院后给予缬沙坦胶囊(80mg,1次/d)联合贝那普利(20mg,1次/d)降压降尿蛋白及调脂等对症支持治疗。院外继续口服上述药物治疗,2个月后24h尿蛋白定量为1.04g,复查心脏彩超示双室肥厚、左室舒张功能减低(Ⅰ级),给予沙库巴曲缬沙坦钠片100mg,2次/d改善心功能,目前该患者仍在我院门诊随访中,随着半乳糖苷酶在我国的上市,后期考虑给予半乳糖苷酶进行治疗。
2 讨论
法布雷病(Fabry disease,Fabry病)又称“Aderson-Fabry病(Aderson-Fabry disease,AFD)”是一种罕见的X染色体连锁性溶酶体贮积症(LSDs)。主要由位于X染色体长臂中的Xq22编码的α-半乳糖苷酶(α-Gal A)的GLA基因发生突变,使得α-Gal A的活性下降或功能丧失,导致溶酶体内的鞘糖脂,尤其以神经酰胺三己糖苷(Gb3)为主无法正常代谢,渐进性的沉积于人体各组织及器官如皮肤、心脏、肾脏、神经系统等,从而造成机体产生一系列渐进性损害[1-3]。患者可以出现四肢神经性疼痛、少汗、皮肤血管角质瘤(以坐浴区多见)、心室肥厚、脑卒中、蛋白尿、严重者肾功能不全等[4]。有数据表明,该病在国外的发病率为1/40 000~1/170 000,在国内尚无人群患病率的统计数据,上海交通大学医学院附属瑞金医院曾报道,在终末期肾衰透析患者中此病的患病率可达0.12%,在意大利,新生儿筛查的发病率为1∶3 100,而在台湾,这一比率可达1∶1 500[1,4]。这表明这一疾病的诊断可能被大多数人忽视,主要与其多样的临床表型相关,目前已知的GLA基因突变已经有1 000多种,基因型与表型之间的关系极其复杂,因为同一基因表型可能导致不同的临床表现,因此该病在临床上的误诊率及漏诊率均较高。
已知的Fabry病主要有经典型和迟发型两种临床表型。经典型主要发生在α-Gal A活性低于1%的男性中,迟发型主要发生在α-Gal A活性高于1%的女性和男性中。由于Fabry病为X连锁性疾病,通常情况下,男性杂合子表现更为严重,女性起病一般较晚,表现更多样化,进展也更为缓慢。经典型的诊断标准为GLA基因发生突变、α-Gal A≤标准值的5%、出现一个或多个Fabry病的经典表现如少汗、肢端疼痛、感觉异常、血管角质瘤等[5-6]。在仅有GLA基因发生突变时,而没有上述经典型的临床表现时归于迟发型。
在经典的亚型中血管内皮细胞中会有明显的鞘糖脂沉积,出现皮肤血管角质瘤、肢端感觉异常、少汗症等。随着疾病的进展将会出现心脏和肾脏的受累,出现心室肥厚、蛋白尿等。值得注意的是,肾脏的表现一般与残存酶活性相关,国外曾有一项研究表明,酶活性低于1%时,在22岁便可诊断为该病,当酶活性在1%~7%时,这一诊断年龄可推迟至47岁[6]。此外,Gb3可以累及几乎所有的肾脏细胞中,如内皮细胞、系膜细胞、足细胞以及肾小管细胞,尤以足细胞为主,因此Fabry病可导致肾小球、血管、肾小管性疾病。蛋白尿的发生主要与Gb3沉积于足细胞、系膜细胞和内皮细胞,最终导致基底膜增厚和肾小球硬化有关[6-8]。足细胞是终末分化的细胞,意味着不可再生,当足细胞受损甚至是丢失时将会导致蛋白尿的加重,严重时发展为慢性肾脏病。
Fabry病对肾组织的累及程度主要以肾活检来确诊,光镜下可出现足细胞的空泡化、泡沫样改变,电镜下出现髓样小体或斑马体是Fabry肾病的典型病理表现[7]。在40%~60%的Fabry病患者中,还会出现心脏受累包括左心室肥厚、心肌缺血、心律失常等,收缩功能将在很大程度上会保留,主要以舒张功能受损为主[8]。
本例患者在入院时心脏彩超提示:左心室肥厚、左室收缩功能正常,在出院2个月后随诊时心脏彩超示:双室肥厚、左室舒张功能减低(Ⅰ级),这与上述Fabry病对心脏受累的特点相契合。此外,该患者并没有出现经典的临床表现及体征,也无相关阳性家族史,院外6年的治疗期间仅存在单纯的蛋白尿,入我院再次行肾组织活检后报告提示高度怀疑Fabry病。随后进行了α-半乳糖苷酶活性的测定及基因检测,其c.902G>A变异在男性中通常会导致迟发型Fabry病,通常在成年期出现心律失常、左心室肥厚(LVH)或蛋白尿以及肾功能不全导致肾衰竭。故结合上述临床病史、临床表现及相关检查结果该患者最终明确诊断为迟发型Fabry病。经给予降压、降尿蛋白及调脂等对症治疗2个月后尿蛋白较入院时减轻,在后期的随访过程中,复查心脏彩超出现双室肥厚、左室舒张功能减低,给予沙库巴曲缬沙坦钠片改善心功能,随着该病纳入医保目录,后期拟给予半乳糖苷酶进行特异性治疗。由于Fabry病会导致多系统改变,其临床表现缺乏特异性,因此临床误诊率极高。当临床病例中出现原因不明的蛋白尿或慢性肾脏病时我们需要考虑到这一疾病的可能,同时临床工作中肾组织活检术将对我们诊断一些原因不明性肾病具有重要的指导意义。
猜你喜欢
数字海洋与水下攻防(2021年2期)2021-05-08
中成药(2018年5期)2018-06-06
中成药(2017年8期)2017-11-22
米娜·女性大世界(2016年8期)2016-08-17
Coco薇(2016年5期)2016-06-03
系统工程与电子技术(2016年2期)2016-04-16
船海工程(2015年4期)2016-01-05
爆笑show(2015年9期)2015-10-24
天然产物研究与开发(2014年3期)2014-04-27
计算物理(2014年1期)2014-03-11
