
基于ProtBert预训练模型的HLA-Ⅰ和多肽的结合预测算法
2023-05-21 03:53:38周丰丰张亚琪
吉林大学学报(理学版) 2023年3期
周丰丰,张亚琪
(1.吉林大学 计算机科学与技术学院,长春 130012; 2.吉林大学 符号计算与知识工程教育部重点实验室,长春 130012)
Ⅰ类人类白细胞抗原(HLA-Ⅰ)是位于人体细胞表面的主要组织相容性复合体(MHC)蛋白,具有启动特异性免疫应答及提呈内源性抗原等作用.HLA-Ⅰ分子可识别位于癌细胞表面的肿瘤抗原肽,但仅可与小部分抗原肽特异性结合,形成呈递给T细胞受体的肽-HLA复合物,若T细胞抗原受体(TCR)能识别癌细胞表面上的肽-HLA复合物,则毒性T淋巴细胞会破坏癌细胞,从而帮助免疫系统在识别病原体方面发挥关键作用.因此,研究多肽与特定的HLA-Ⅰ分子的结合机制对癌症免疫治疗和基于蛋白质的疫苗及药物开发具有重要意义.
HLA-Ⅰ分子具有高度多态的特性,根据数据库IMGT/HLA统计,目前已有超过23 000个Ⅰ类HLA等位基因,它们主要由HLA-A、B、C位点编码,因此对于HLA-Ⅰ和多肽的结合预测模型分为两类: 特异性模型和泛特异性模型.特异性模型即针对每个HLA-Ⅰ等位基因训练一个对应的预测模型,如NetMHC[1]和SMM[2]等; 泛特异性模型则是对所有同种异型的HLA-Ⅰ分子训练一个通用预测模型,如NetMHCPan[3]等.目前,仍有很多HLA-Ⅰ等位基因只具有少量的已知结合肽,由于泛特异性模型使用多个等位基因的数据进行训练,不会被训练数据集的大小限制,因此本文提出的ProHLAⅠ框架是一种泛特异性模型.目前,主流的预测方法有基于序列评分函数的方法和基于机器学习的方法两种.用于氨基酸序列预测问题的序列评分函数已有许多种,例如氨基酸频率评分[4-5]、基于WebLogo的熵信息[6]等.MHCflurry1.2.0[7]和HLA-CNN[8]等使用了机器学习和深度学习算法进行模型的搭建,基于原始的氨基酸序列生成更高维度的特征,从而实现预测任务.
蛋白质序列可被视为一种生命体语言,虽然蛋白质的结构和功能是动态的和上下文相关的,但其仍由潜在的氨基酸序列定义.像句子是由若干单词组成一样,蛋白质由一串氨基酸序列定义,目前已知的氨基酸共20余种,不同氨基酸的排列组合会形成不同功能的蛋白质.因此,从信息论的角度,蛋白质的信息被包含在它的序列中[9].鉴于上述生命体语言和文本语言的共性,深度学习算法已逐渐从自然语言处理领域(NLP)迁移至生物信息学[10-11],UniRep[12]将深度学习应用于未标记的氨基酸序列,从蛋白质序列中提取语义丰富的统计表示特征,并在下游任务中表现出优越性.TAPE[13]在5个与蛋白质相关的半监督学习任务中验证了基于NLP的蛋白质嵌入编码方式可更好地捕捉生物序列中的信息.预训练语言模型BERT(bidirectional encoder representations from transformers)[14]一经提出就在11项NLP任务中取得了最先进的结果,Elnaggar等[15]使用大量蛋白质序列数据对原始的BERT模型进行了进一步微调,提出了蛋白质预训练模型ProtBert.本文基于ProtBert提出一种新的HLA-Ⅰ和多肽的结合预测算法ProHLAⅠ,整合预训练ProtBert模型、BiLSTM序列编码能力和注意力机制,提取氨基酸序列的上下文信息生成高维度特征向量.实验结果表明,将NLP领域中的文本语言模型迁移至蛋白质序列预测问题上,可有效提高预测的准确性.
1 算法设计
1.1 蛋白质预训练模型ProtBert
1.1.1 BERT
BERT是一种基于Transformer模块的自然语言处理领域预训练模型.BERT采用多个双向Transformers编码模块组成,每个编码模块由Self-Attention子层和Feed Forward子层组成,在每个子层后进行残差连接和层归一化操作,其结构如图1所示.
BERT由预训练和微调两个阶段任务组成.预训练阶段是利用大型语料库对序列语言双向编码模型进行无监督训练.定义了两种全新的无监督预测任务优化网络参数,分别是基于Mask机制的语言预测任务(简称Masked LM)[14]以及下一句预测任务,微调阶段则针对具体的下游任务对与训练模型进行微调.

图1 Transformers Encoder模型Fig.1 Transformers Encoder model
1.1.2 ProtBert
蛋白质预训练模型ProtBert[15]使用来自UniRef100[16]和BFD[17]的21亿个蛋白质序列对原始的BERT模型进行进一步训练,将蛋白质序列解释为句子,将其组成成分氨基酸解释为单个单词,将模型从文本语言映射到生命体语言.与原始的BERT模型相比,ProtBert将组成模型的Transformer Encoder模块层数提高至30层,以便在受监督的下游任务中获得更好的性能,同时调整了模型的训练策略,使该模型能在使用较大batch_size的同时首先从较短的序列中提取有用的特征,从而提高了对较长序列的训练效率,进而提高了整体训练效率.Elnaggar等[15]将ProtBert作为特征提取器在3个不同的下游任务(二级结构预测、定位预测以及膜或非膜蛋白的分类)中证明了将BERT迁移至蛋白质序列问题上的鲁棒性.
1.2 BiLSTM神经网络
循环神经网络(recurrent neural network,RNN)广泛用于时间序列数据类型内在模式的建模,但经典RNN网络在处理长序列时具有长期依赖的问题.LSTM(long short-term memory)通过“门”结构控制网络中信息的传输,使网络可选择性地记住或遗忘某些信息,有效解决了长序列在训练过程中丢失较远信息的问题.BiLSTM(bi-directional long short-term memory)由前向和后向两个LSTM神经网络组成,可以同时捕捉序列从前向后和从后向前的双向语义依赖信息,解决了LSTM无法编码从后向前语义信息的缺陷.
1.3 Attention机制
深度学习领域中的注意力(Attention)机制模仿人类观察行为的过程,将注意力聚焦在重点信息区域,从而获取更多的相关信息,减少无关信息的干扰.目前,注意力机制在多领域应用广泛,如机器翻译、自然语言处理、计算机视觉和蛋白质序列建模等.Attention机制在神经网络处理大量输入特征时,对不同的特征赋予不同的权重,使与任务相关的输入信息参与下一步网络的计算,从而提高了神经网络处理信息的能力.
Attention机制的模型结构如图2所示,其中Query向量,Key向量和Value向量在不同的具体任务中会根据输入序列初始化,计算过程如下:

图2 Attention机制模型Fig.2 Attention mechanism model
步骤1) 利用相似度函数,计算每个Key向量与Query向量的相似度,作为权重;
步骤2) 使用Softmax函数对权重进行归一化操作;
步骤3) 将每个Value向量与对应的权重加权求和,得到Attention的输出.
1.4 ProHLAⅠ网络
ProHLAⅠ网络包括预处理模块、词嵌入的预训练模块、BiLSTM模块、注意力模块和分类模块,其结构如图3所示.
预处理模块的输入为原始HLA-Ⅰ分子和多肽对,由下列4个步骤组成:
1) 编码.采取NetMHCPan中提出的HLA-Ⅰ伪序列转换方式将HLA-Ⅰ分子转换为长度为34的伪序列;
2) 补全.对于长度小于15的多肽,氨基酸“X”会被补至序列末尾,使多肽长度统一为15;
3) 连接.将HLA-Ⅰ伪序列与补全后的多肽序列连接,生成长度为49的HLA-多肽序列;
4) 将HLA-多肽序列首尾添加[CLS]和[SEP]标志以符合ProtBert模型的输入格式.

图3 ProHLAⅠ框架Fig.3 ProHLAⅠ framework
最终预处理模块的输出是长度为51的氨基酸序列.词嵌入的预训练模块利用ProtBert语言模型提取预处理后的氨基酸序列中的高维特征,并将初步提取的特征作为BiLSTM编码模块的输入,BiLSTM编码模块进一步学习氨基酸序列中的上下文依赖关系,然后注意力模块将BiLSTM层的输出加权融合得到最终的1 536维特征向量,通过全连接神经网络和Softmax归一化得到HLA-Ⅰ分子和多肽的结合预测结果,最后整个模型的输出为一个一维向量,标识预测结果.
2 实 验
2.1 数据集
实验数据集选取DeepHLApan[18]中整理的来自数据库IEDB(http://www.iedb.org/)的81个HLA-Ⅰ等位基因和长度为8~15的多肽序列.共有437 077个HLA-多肽对参与实验模型训练,其中正样本和负样本的比例为1∶1,本文按照7∶2∶1将数据集均衡地分为训练集、验证集和测试集,用于模型调优和消融实验.
从IEDB的每周基准测试集中下载11个子数据集作为独立测试集1,并从IEDB上下载其他7种预测算法在上述数据集上的预测性能用于模型评估.采用文献[19]中的独立数据集作为独立测试集2,该数据集由15个子数据集组成,且从未被开发工具用于模型训练等.
2.2 评价指标
本文将HLA-Ⅰ和多肽的结合预测规定为二分类任务.通过3个性能指标ROC曲线下面积(AUC)、准确率(ACC)和F1分数(F1-score)对模型预测结果进行评估,计算公式分别为

(1)
其中TP和FN分别表示真阳性和假阴性的数量,TN和FP分别表示真阴性和假阳性的数量,Precision表示精确率,Recall表示召回率.AUC是一个用于衡量分类器性能的指标,其值越大,分类器性能越优.
2.3 参数设定
实验的初始学习率设为5×10-5,训练轮次epoch为3,batch size设为16,实验中使用dropout防止过拟合,dropout率为0.1.
2.4 消融实验
2.4.1 batch size参数评估
为确定最优的batch size参数,本文在仅适用ProtBert模型做特征提取器,后接分类模块的前提下,分别设batch size为16,32和64进行对比实验,在验证集上得到的性能指标结果如图4所示.由图4可见,当batch size设为16时,ACC,AUC和F1分别得到了最优结果.因此,所有后续实验batch size参数均设为16.
2.4.2 ProtBert结合模型评估
采用多模块结合的方式预测HLA-Ⅰ和多肽的结合,实验中评估4种不同的模块结合方式以达到最好的预测结果,分别为ProtBert,ProtBert+BiLSTM,ProtBert+CNN及本文提出的ProHLAⅠ框架,在验证集上得到的性能指标结果如图5所示.由图5可见,对ProtBert模型融合其他模块可提升模型的分类性能,本文提出的ProHLAⅠ框架在3个评价指标下均达到了最优.因此,本文的最终模型采取了ProHLAⅠ框架结构.

图4 不同batch size参数下的性能评估结果Fig.4 Performance evaluation results under different batch size parameters
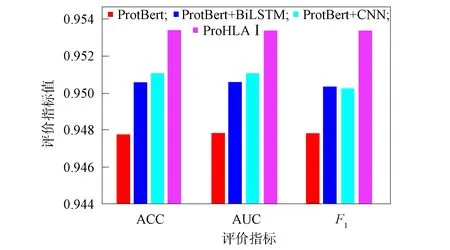
图5 不同结合模型的性能评估结果Fig.5 Performance evaluation results of different combination models
2.5 实验结果与分析
表1为使用独立测试集1的11个子数据集,对本文提出的ProHLAⅠ与其他7种HLA-Ⅰ和多肽结合预测算法(NetMHCpan 4.0,SMMPMBEC,IEDB Consensus,NetMHCcons,PickPocket,MHCflurry 1.2.0和DeepHLApan)在AUC指标下的对比结果.

表1 不同预测算法对独立测试集1的AUC指标对比结果
由表1可见,本文提出的预测算法在11个子数据集的6个中均得到了最高的AUC值,是8种预测算法中性能表现相对最稳定和最优的.即使对于所有预测算法的AUC值均不超过0.7的子数据集中,本文模型仍可在多数数据集上取得最优性能,从而证明了仅依赖氨基酸序列的上下文关系进行特征提取的方式对那些本身性能表现较差的数据会有更突出的预测效果.
表2为使用独立测试集2的15个子数据集,对本文提出的模型与其他6种结合预测算法(Pickpocket,MixMHCpred,NetMHCpan 4.0,NetMHCcons,NetMHCstabpan和DeepHLApan)在AUC指标下的对比结果.由表2可见,所有算法在这些数据集上都取得了相似的性能,本文提出的ProHLAⅠ框架在其中的14个子数据集上的AUC指标值超过了0.990,并在其中10个子数据集上得到了最优的性能.

表2 不同预测算法对独立测试集2的AUC指标对比结果
通过在两组共26个独立子数据集上的性能比较,本文算法在其中的16个子数据集上均有最优的性能,在所有预测算法中是预测性能最稳定的,因此本文提出的ProHLAⅠ框架可获得与最新的HLA肽结合工具相当、甚至更优的性能.
综上所述,本文针对HLA-Ⅰ结合肽预测问题,提出了一种基于NLP算法的模型,用于氨基酸序列的特征提取.本文研究不依赖序列评分函数,仅使用HLA-Ⅰ分子和多肽序列的自身信息构建特征.通过预训练ProtBert模型初步提取序列中的重要特征,采用BiLSTM网络进一步提取氨基酸序列上下文特征,基于注意力机制对提取的特征加权融合得到最终用于模型分类的特征向量.通过在两组独立测试集上的验证结果表明,本文提出的预测模型均取得了最优性能,证明了整合自然语言处理领域的ProtBert预训练模型、BiLSTM序列编码功能和注意力机制编码氨基酸序列的必要性,进而表明了将自然语言处理算法应用于蛋白质生命语言上的可行性[20-21].
猜你喜欢
数学年刊A辑(中文版)(2020年2期)2020-07-25 02:04:44
数学物理学报(2019年6期)2020-01-13 06:08:16
中国洗涤用品工业(2019年4期)2019-05-11 09:27:18
中成药(2018年1期)2018-02-02 07:20:05
现代园艺(2017年13期)2018-01-19 02:28:09
数学物理学报(2017年5期)2017-11-23 07:51:31
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
动物医学进展(2015年10期)2015-12-07 05:46:19
药学与临床研究(2015年4期)2015-06-05 11:35:54
科学中国人(2015年16期)2015-02-28 09:14:02
