
Novel homozygous ADAMTS17 missense variant in Weill-Marchesani syndrome
2023-05-15 09:20NaMiaoYaoZhangJinYingLiaoLinZhouJiCaiHeRongQinYangXuYangLiuLiTang
Na Miao, Yao Zhang, Jin-Ying Liao, Lin Zhou, Ji-Cai He, Rong-Qin Yang, Xu-Yang Liu,Li Tang
1Department of Ophthalmology, West China Hospital of Sichuan University, Chengdu 610041, Sichuan Province,China
2Department of Ophthalmology, First People’s Hospital of Liangshan Yi Autonomous Prefecture, Xichang 615306,Sichuan Province, China
3Xiamen Eye Center, Xiamen University, Xiamen 361011,Fujian Province, China
4Department of Ophthalmology, Shenzhen People’s Hospital,the 2nd Clinical Medical College, Jinan University, Shenzhen 518040, Guangdong Province, China
Abstract
INTRODUCTION
Weill-Marchesani syndrome (WMS) is a rare hereditary connective tissue disease characterized by ocular problems, including microspherophakia, ectopia lentis, high myopia, and secondary glaucoma.Other symptoms include short stature, brachydactyly, joint stiffness, and occasional cardiac defects.The syndrome usually involves a family history or a history of consanguinity between parents or close relatives.WMS can show autosomal dominant or autosomal recessive inheritance.The most common pathogenic genes linked to WMS are theFBN1,LTBP2,ADAMTS10 and ADAMTS17genes[1-8].Regardless of which of these genes is varied in WMS patients, the disease features appear to be similar.Here, we report a new homozygous variation inADAMTS17(MIM*607511, ADAM metallopeptidase with thrombospondin type 1 motif, 17) leading to autosomal recessive WMS in a Chinese family.
SUBJECTS AND METHODS
Ethical ApprovalThis study followed the principles of the Declaration of Helsinki, and it was approved by the Ethics Committee of West China Hospital of Sichuan University(2019, No.53).All subjects fully understood the purpose of the study and provided written informed consent.
Patient Ascertainment and Clinical AssessmentThe proband and other family members were invited to our hospital for a detailed medical history inquiry and complete physical and ophthalmic examination, including height measurement,ECG, hands and feet X-ray, cardiac ocular ultrasound, bestcorrected visual acuity testing, intraocular pressure (IOP)measurement (Goldmann tonometry), slit lamp examination,fundus examination, gonioscopy, ultrasound biomicroscopy(UBM), corneal endothelial cell count, B-ultrasound, ocular biometry measurement, visual field and anterior segmentoptical coherence tomography (AS-OCT, Cirrus HD-OCT 4000) testing.
Whole-Exome Sequencing and SANGER SequencingPeripheral venous blood (3 mL) was collected from each family member for DNA extraction and genotyping.The exons and adjacent splicing regions (approximately 20 base pairs) of the target gene and the full length of the mitochondrial genome were captured and enriched by probe hybridization.The enriched genes were quality controlled and sequenced using a high-throughput sequencer.According to the selected mutation sites, the consolation of other family members was verified.
Bioinformatic AnalysisSequences of the wild-typeADAMTS17gene and encoded protein were downloaded from the National Center for Biotechnology Information (NCBI,https://www.ncbi.nlm.nih.gov/), and mutated amino acids in theADAMTS17protein were changed manually.The online Cobalt tool (https://www.ncbi.nlm.nih.gov/tools/cobalt) was used to alignADAMTS17proteins from different species to determine whether a mutated position was conserved.The potential functional implications ofADAMTS17mutations were predicted using PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://provean.jcvi.org/).The structures of mutantADAMTS17proteins were modeled based on homology with the wild-type protein in PyMOL (https://swissmodel.expasy.org).
RESULTS
Family CharacteristicsThis case study involved a family of nine members, all Han Chinese, from three generations identified at West China Hospital of Sichuan University.Three of the individuals were diagnosed with WMS, including one male (Ⅳ‐3) and two females (Ⅳ‐2 and Ⅳ‐5; Figure 1).The proband was a 22‐year‐old patient Ⅳ‐3, who was referred to our clinic with complaints of decreased vision accompanied frequently by eye soreness in both eyes for 5y.He was diagnosed with bilateral secondary angle-closure glaucoma.His sister Ⅳ‐5 had high myopia in both eyes that was not corrected by glasses and complained of eye soreness after reading for long periods.The other sister, ⅠⅤ‐2, had poor visual acuity but never visited the hospital before.Their parents (III-1 and III-2) were in consanguineous marriage.The inheritance pattern of WMS in this family was autosomal recessive.The family pedigree is depicted in Figure 1.

Figure 1 Pedigree showing inheritance of WMS in a Han Chinese family with a history of consanguineous marriages Black identifies individuals diagnosed with the syndrome.The arrow marks the proband, IV-3.WMS: Weill-Marchesani syndrome.
The height of the three affected siblings was significantly lower than that of other family members.The other systemic abnormalities included brachydactyly but had no anomalies in the cardiovascular system (Table 1, Figure 2).Ophthalmic examination revealed high myopia, a very shallow anterior chamber, microspherophakia with stretched zonules, lens subluxation and angle closure glaucoma (Figure 3).Their clinical findings are summarized in Table 1.Based on these findings, the proband and his sister (Ⅳ‐5) underwent surgeries for removal of the dislocated lens and implantation of an intraocular lens (IOL).Capsular bag stabilization was accomplished with capsular hooks during the procedure, and a capsular tension ring was inserted prior to IOL implantation in the capsular bag.Following surgery, visual acuity significantly improved, and the IOP was normalized with a stable, deepened anterior chamber.
GenotypingA homozygous missense variation c.2983C>Tp.Arg995Trp) was detected in theADAMTS17gene in all members of the family with the disease.Ⅰndividuals Ⅲ‐1, Ⅲ‐2,Ⅳ‐1, Ⅳ‐6, and Ⅴ‐1 were heterozygous for the same mutation.One individual was homozygous for the wild-type allele.This mutation changed C2983 to T in the cDNA, resulting in an Arg995Trp substitution in the protein (Figure 4).

Figure 2 Clinical features of patients A: Images of the hands of proband IV-3 and sister IV-5 showing short fingers; B: X-ray of both hands showing a shorter phalanx normal joint.

Figure 3 Ophthalmological characterization of the proband (IV-3) A:Slit-lamp examination revealed a very shallow anterior chamber in both eyes; B: Anterior segment photographs, after dilatation of the pupil,showing a small spherical lens and a gold ring, which was caused by the reflection of light from the 360° periphery of the small crystalline globular lens with stretched zonules, resulting in the subluxation of the lens; C: UBM showed a shallow anterior chamber in both eyes,a forward-shifted lens iris, and a spherical anterior surface on the lens; D: The AS-OCT, before (a) and after (b) dilatation of the pupil(left eye), showed a very shallow anterior chamber and a subluxated small spherical lens, and the zonules were still not relinquished; E:Fundoscopy showed advanced glaucomatous cupping in both eyes.

Figure 4 Partial sequencing of the ADAMTS17 gene in WMS patients of the family A homozygous missense mutation (c.2983C>T: p.Arg995Trp, red arrow) was found in the ADAMTS17 gene of all patients in this family.WMS: Weill-Marchesani syndrome.
Bioinformatics AnalysisThe 995Arg position of theADAMTS17protein (NP_620688.2) is extremely conserved among different species, such as humans (Homo sapiens),mice (Mus musculus), zebrafish (Danio rerio), frogs (Xenopus tropicalis), rhesus monkeys (Macaca mulatta), rats (Rattus norvegicus), chickens (Gallus gallus), goats (Capra hircus),rabbits (Oryctolagus cuniculus) and hamsters (Cricetulus griseus), according to the alignment of protein sequences shown in Figure 5.The PolyPhen-2 analysis results showed that the missenseADAMTS17variant (c.2983C>T: p.Arg995Trp)was predicted to be PROBABLY DAMAGING with a score of 1000.The PROⅤEAN score was ‐4.928, deleterious,according to the PROⅤEAN prediction results.Two protein functional prediction tools obtained the same harmful results,indicating that this variant should be pathogenic.Furthermore,the structural prediction results of PyMOL showed that there might be no large structural change between the mutated and wild-type ADAMTS17 proteins.However, the charged and hydrophobic status should be changed at the 995 position, as shown by the black arrows in Figure 5.
DISCUSSION
Ⅰn this study, we identified a novel homozygousADAMTS17missense variant (c.2983C>T: p.Arg995Trp) associated with WMS.Three of nine siblings in the family were diagnosed with WMS.

Figure 5 Bioinformatics analysis A: Protein sequence alignment at the 995 position (red box) among different species; B: Structural modeling by PyMOL showing charged and hydrophobic status.a: Charged wild type; b: Charged mutated protein; c: Hydrophobic wild type; d:Hydrophobic mutated; Black arrow: Mutated position.

Table 1 Clinical features of the affected individuals in the family
The proband and his two affected sisters presented with angle closure glaucoma.The proband and one of his sisters (Ⅳ‐2)showed optic atrophy.Another sister (Ⅳ‐5) did not exhibit obvious optic atrophy.The reason for the inconspicuous optic atrophy was that she was the youngest and had a short onset time.
To prevent further injury of the optic nerve and improve visual acuity, two affected individuals (proband and Ⅳ‐5) underwent surgeries to remove the dislocated lens and implant the IOL.In this case, to remove the lens more safely and with less risk of further zonular damage, iris retractors were used to temporarily support and stabilize the capsular bag; then, a capsular tension ring was inserted prior to IOL implantation in the bag.The surgeries were uneventful, and the patients were doing well.
PathogenicADAMTS17variants were first reported in 2009[3].To date, eight variations in the humanADAMTS17gene have been reported, including a nonsense mutation (c.1051A>T),a missense mutation (c.760C>T), c.1027A>G, splice-site mutations (c.873+1G>T and c.1721+1G>A), indels (including c.2458_2459insG, c.652delG and a 106.96 Kb deletion containing exon 1–3 regions.Our study increases the number of variations of this gene to nine (Table 2)[1,3,9-12].
None of the three individuals with WMS in our study showed joint stiffness or cardiac abnormalities, consistent with the lack of such symptoms in other reports of WMS associated withADAMTS17variants (Table 2).Review the literature,all the patients had ocular abnormalities and short stature,but only a few patients had brachydactyly, joint stiffness, and cardiac abnormalities.Cardiac abnormalities were reported in only 3 of 18 patients.One of the patients had concomitant tachycardia, mitral valve dysplasia, and cardiomyopathy, and two patients had mitral valve dysplasia[9]Our study adds tothe range ofADAMTS17polymorphisms in humans (Table 2).Further studies should examine whether WMS associated withADAMTS17variants represents a different subtype of the disease or simply a milder manifestation of the typical disease phenotype.
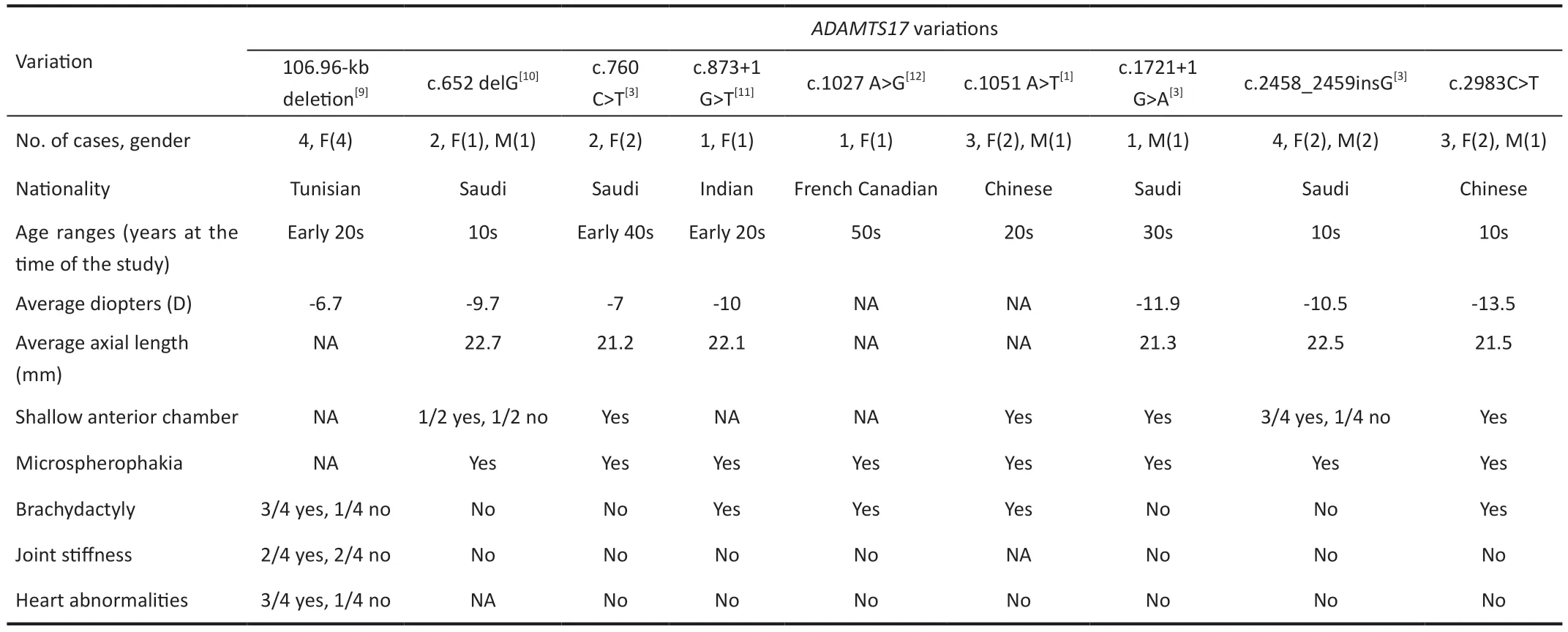
Table 2 Summary of clinical phenotypes of known ADAMTS17 variations linked to WMS
ADAMTS17, located at position 15q26.3 (MIM 613195),encodes a member of the ADAMTS family of secreted metalloproteases, which are involved in extracellular matrix(ECM) formation, remodeling and degradation[13].ADAMTS17,which in the eye is expressed mainly near the crystal epithelium, appears to promote ECM formation, especially the assembly of fibrillin microfibrils[2,11].ADAMTS17appears to stabilize zonular microfibrils[14]; therefore, deficiency in the protein may destabilize microfibrils, causing abnormalities in the lens zonule[12,14].Consistent with this idea, polymorphisms inADAMTS17have been linked to primary open angle glaucoma and ectopia lentis in dogs[15-16].Future studies should explore the role ofADAMTS17polymorphisms in the pathogenesis of WMS.
Short stature is a distinct feature of WMS, andADAMTS17polymorphism in humans was linked to height[17], as have variations in copy number at a locus near this gene[18-19].ADAMTS17polymorphism has also been linked to height in dogs[15-16], suggesting thatADAMTS17plays an important role in normal growth and development.
In this study, we analyzed the gene variation in the adjacent splicing region of the whole exome of the proband and revealed a homozygous missense variant c.2983C > T: p.Arg995Trp on theADAMTS17gene, which segregated with the phenotype in the pedigree.This variant site is in three TSP1 repeats, which may destabilize zonules by affecting ECM and microfibril assembly, causing zonular abnormalities, spherical crystals, or crystal subluxation.
ACKNOWLEDGEMENTS
Foundations:Supported by The Cadre Health Research Program of the Sichuan Province (No.2023-119); Sichuan Science and Technology Program (No.2021YFS0213).
Conflicts of Interest:Miao N,None;Zhang Y,None;Liao JY,None;Zhou L,None;He JC,None;Yang RQ,None;Liu XY,None;Tang L,None.
 International Journal of Ophthalmology2023年5期
International Journal of Ophthalmology2023年5期
- International Journal of Ophthalmology的其它文章
- Analysis of retinal arteriolar and venular parameters in primary open angle glaucoma
- ldentification and functional analyses of a novel FOXL2 pathogenic variant causing blepharophimosis, ptosis,and epicanthus inversus syndrome
- Protective effects of ferulic acid against ionizing radiation-induced oxidative damage in rat lens through activating Nrf2 signal pathway
- Cost analysis of childhood glaucoma surgeries using the US Medicaire allowable costs
- Predicting the prognosis of primary orbital lymphoma by clinical characteristics and imaging features
- Guidelines from an expert panel for the management of diabetic macular edema in the Malaysian population