
利多卡因对大鼠缺血-再灌注心律失常的影响及机制探讨*
2023-05-08 10:22王静唐玲肖婷
中国心脏起搏与心电生理杂志 2023年2期
王静 唐玲 肖婷
研究表明,缺血再灌注可引发心肌组织强烈炎症及氧化应激,并导致过度自噬,造成心肌细胞凋亡,是导致心律失常的主要病理基础,减轻炎症、自噬及氧化应激,是缺血-再灌注心律失常的一种有效治疗策略[1-3]。利多卡因是一种钠通道阻滞剂,可有效改善急性心肌梗死(简称心梗)患者心功能,减轻其心律失常症状[4],但其药理机制目前尚未见明确报道。磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)信号可调节细胞生长、凋亡、自噬等生理过程,在缺血-再灌注心律失常的发病机制中起到关键的调控作用。激活PI3K/AKT 信号,可减少炎症因子的产生,降低氧化应激水平,抑制心肌细胞凋亡,改善心律失常症状[5-6],并通过抑制过度自噬而减轻心肌缺血-再灌注损伤[7]。研究表明利多卡因能通过增强PI3K/AKT 信号通路表达降低心力衰竭大鼠炎性因子合成释放,抑制其心肌细胞凋亡,保护心脏功能[8],因而激活PI3K/AKT 信号可能是利多卡因治疗缺血-再灌注心律失常的药理机制。笔者探究利多卡因对缺血-再灌注心律失常的影响及其机制。
1 材料与方法
1.1 实验动物SPF 级SD 大鼠购自广州锐格生物科技有限公司[SCXK(粤)2021-0059],体重均为190~230 g,雄性,6周龄左右,在本院实验动物中心分笼饲养,动物房温度设在23~25℃,湿度设在50%~60%,并严格按照«中华人民共和国实验动物管理条例»进行动物管理及实验操作。
1.2 实验试剂盐酸利多卡因注射液(规格1.8 ml:36 mg,国药准字H20013390)购自黑龙江哈尔滨医大药业有限公司;PI3K/AKT 信号抑制剂LY294002(纯度:99.87%,货号HY-10108)购自美国MCE 公司;丙二醛(malondialdehyde,MDA)测定试剂盒(货号A003-1-2)、TUNEL 细胞凋亡检测试剂盒(货号G001-1-1)、BCA 蛋白质定量检测试剂盒(货号C503021)、谷胱甘肽过氧化物酶(glutathione peroxidase,GSH-Px)测定试剂盒(货号A005-1-2)、活性氧(reactive oxygen species,ROS)测定试剂 盒(货 号 E004-1-1)、RIPA 裂 解 液(货 号C500005)、诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)试剂盒(货号H372-1)、前列腺素E2(Prostaglandin E2,PGE2)酶联免疫试剂盒(货号D751014)均购自南京建成生物工程研究所有限公司;兔源Anti-LC3一抗(货号ab179463)、兔源Anti-AKT 一 抗(货 号ab179463)、兔 源Anti-Beclin1一抗(货号ab138364)、兔源Anti-p-PI3K 一抗(货 号ab138364)、兔 源 Anti-Bax 一 抗(货 号ab151549)、兔源Anti-PI3K 一抗(货号ab151549)、兔源Anti-caspase-9一抗(货号ab38449)、兔源Anti-p-AKT 一抗(货号ab38449)、羊抗兔二抗(货号ab150077)、兔源Anti-GAPDH(货号ab181602)一抗均购自美国Abcam 公司等。
1.3 缺血-再灌注心律失常模型制备及分组取SD 大鼠,参照文献[9]制备缺血-再灌注心律失常模型:腹腔注射40 mg/kg的3%戊巴比妥钠溶液,待大鼠进入深度麻醉状态时,将其仰卧位绑缚固定在操作板上,气管插管后,连接小动物呼吸机(型号Kent,肯特企业集团公司)做机械通气,并以十六道生理参数分析记录仪(型号GY-6168B,河南华南医电科技有限公司)观察监视大鼠心电变化,将大鼠前胸脱毛、备皮、消毒后开胸,暴露心脏后,游离肺动脉,于其圆锥旁出针并穿过心肌表层,进行结扎,阻断冠状动脉血流,当大鼠左心室前壁出现紫绀,心电图ST 段显著抬高,表示心肌缺血成功,30 min后去除结扎线,恢复血流灌注2 h即表示造模成功完成,用于后续实验。将盐酸利多卡因注射液、LY294002 均以0.9%氯化钠溶液稀释,得到0.4、0.8 mg/ml的利多 卡 因 药 液[10]、利 多 卡 因(0.8 mg/ml)和LY294002(1.0 mg/ml)的混合药液[11]。
SD 大 鼠 共70 只,用 于 构 建 模 型 大 鼠58 只(7只死亡,3只未达到构模标准),检测左室收缩压(112.62±11.36)mm Hg,心 率(338.14±22.58)次/分,将建模后心肌缺血成功的大鼠随机分为模型组、利多卡因低剂量(4 mg/kg)组、利多卡因高剂量(8 mg/kg)组、利多卡因(8 mg/kg)+PI3 K 抑 制(10 mg/kg)组,每 组12 只,另 取12 只大鼠只开胸暴露心脏,不结扎肺动脉,作为假手术组,心肌缺血成功后各组依组别马上进行给药处理:利多卡因低剂量与高剂量组大鼠分别腹腔注 射10 m L/kg剂 量 的0.4、0.8 mg/m L 利 多 卡因药液;利多卡因+PI3 K 抑制组大鼠以10 m L/kg的剂量腹腔注射利多卡因(0.8 mg/m L)和PI3 K 抑 制 剂LY294002(1.0 mg/m L)的 混 合 药液;假手术组与模型组大鼠腹腔注射等剂量的0.9%氯化钠溶液,每组给药处理1 次后恢复血流灌注。
1.4 心律失常检测及标本采集恢复血流灌注2 h后,以十六道生理参数分析记录仪记录大鼠心电图,打开应用程序,输入组别、鼠号,设置低通滤波为600 Hz,屏速调为100 mm/s,选择1-5通道,连接5组导联线,正极接到大鼠右上肢,负极接到大鼠左下肢,地线接到右下肢,观察并记录大鼠心电变化,获得大鼠心电图,根据心电图波形变化分析得到大鼠心室颤动(简称室颤)持续时间与1 min内室性早搏(简称室早)次数。然后以1.3中方法麻醉大鼠,以注射器采集颈动脉血,4℃下以3000 rpm/min的转速离心15 min,将血清吸出在-80℃保存;颈椎脱臼处死大鼠,开胸取出心脏,以手术剪剪下0.4 g,加入生理盐水冰水浴中匀浆,4℃下以3000 rpm/min的转速离心20 min,将上清吸出保存在-80℃,以手术剪再剪下心肌组织0.9 g,加入RIPA 裂解液冰水浴中匀浆,4℃下以3000 rpm/min的转速离心20 min,将上清吸出在-80℃保存,剩余的心肌组织经包埋后置于液氮中冻成块状,取出放入冰冻切片机(型号FS800,深圳市瑞沃德生命科技有限公司)中切片备用。
1.5 心肌细胞凋亡检测取1.4中的大鼠心肌组织切片,复温后置于预冷后的丙酮中固定,以试剂盒进行TUNEL染色,具体按照试剂盒说明书指导步骤进行,采用光学显微镜(型号DSX100,日本奥林巴斯株式会社)观察封片后的心肌组织着色情况,凋亡细胞被染成棕色或深棕色,拍照后以Image-J软件分析计算出心肌细胞凋亡率=凋亡细胞数/总细胞数×100%。
1.6 血清iNOS、PGE2 及心肌组织氧化应激因子测定取1.4中的颈动脉血清与心肌组织上清液,提前放置在冰水浴中解冻,采用试剂盒中试剂并通过酶标仪(型号2010ELX-808,美国Bio Tek 公司)测量血清iNOS、PGE2水平及心肌组织中氧化应激因子GSH-Px、ROS、MDA 水平,具体按照试剂盒说明书指导步骤进行。
1.7 心肌组织凋亡、自噬与PI3K/AKT通路蛋白表达情况检测 取1.4中的心肌组织蛋白样品液,以BCA 试剂盒测出各组蛋白总浓度后将其调至相同,每组取20μL煮沸变性,以蛋白电泳及转膜系统(型号170-4070,美国Bio-Rad公司)进行电泳与湿转,获得分离开的蛋白,以5%脱脂奶粉封闭其非特异位点,兔源Anti-Bax、Anti-caspase-9、Anti-Beclin1、Anti-LC3、Anti-AKT、Anti-p-PI3K、Anti-PI3K、Anti-p-AKT、Anti-GAPDH 一抗孵育,洗膜,羊抗兔二抗孵育,洗膜,化学发光法显色,拍照,Image-J软件分析定量各蛋白灰度值,经统计后得到其相对表达量。
1.8 统计学分析实验数据均为计量资料,以均数±标准差(±s))表示,采用SPSS 24.0软件进行统计分析,假手术组和模型组之间比较进行t检验;多组间比较进行单因素方差分析,多组间进一步两两比较进行SNK-q检验,以P<0.05为差异有显著性。
2 结果
2.1 各组大鼠心律失常的比较与假手术组相比,模型组室颤持续时间和室早次数显著升高(P<0.05);与模型组相比,利多卡因低剂量与高剂量组大鼠室颤持续时间和室早次数均降低(P<0.05);与利多卡因低剂量组相比,利多卡因高剂量组大鼠室颤持续时间和室早次数降低(P<0.05);与利多卡因高剂量组相比,利多卡因+PI3 K抑制组大鼠室颤持续时间和室早次数升高(P<0.05)。见图1、表1。

图1 各组大鼠心电图检测
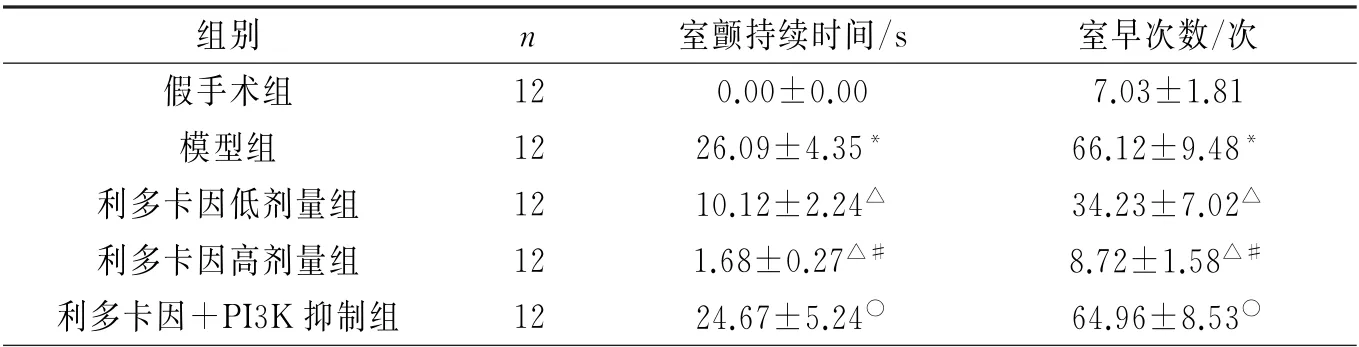
表1 各组大鼠室颤持续时间和室早次数
2.2 各组大鼠心肌细胞凋亡的比较与假手术组相比,模型组心肌细胞凋亡率显著升高(P<0.05);与模型组相比,利多卡因低剂量与高剂量组心肌细胞凋亡率均降低(P<0.05);与利多卡因低剂量组相比,利多卡因高剂量组、心肌细胞凋亡率降低(P<0.05);与利多卡因高剂量组相比,利多卡因+PI3K 抑制组心肌细胞凋亡率升高(P<0.05)。图2、表2。

表2 各组大鼠心肌组织凋亡蛋白表达水平及心肌细胞凋亡率/%
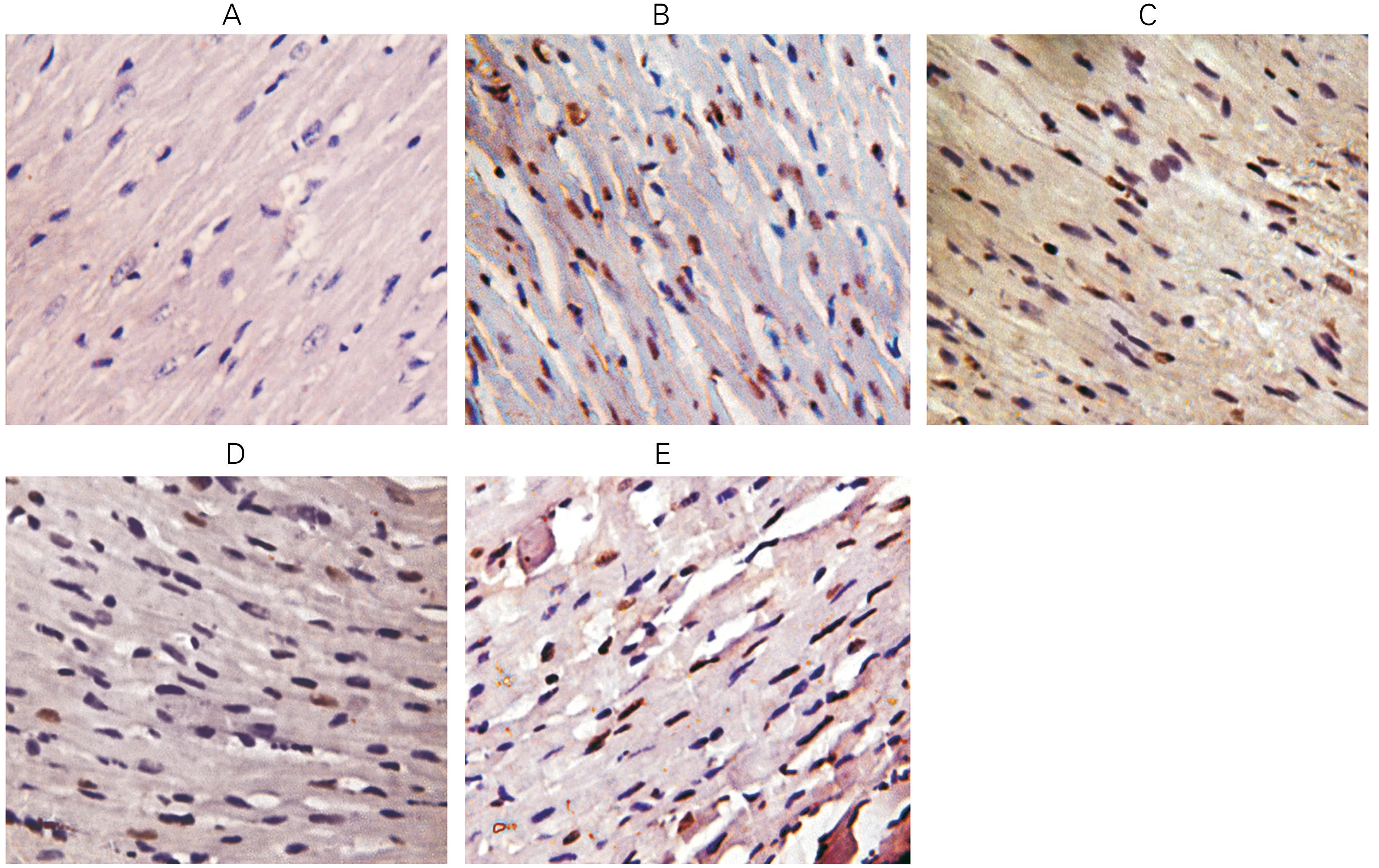
图2 各组大鼠心肌组织TUNEL染色结果(×200)
2.3 各组大鼠心肌组织氧化应激与炎症的比较与假手术组相比,模型组心肌组织GSH-Px水平显著降低(P<0.05),心肌组织ROS及MDA 水平、血清炎症因子iNOS 及PGE2水平显著升高(P<0.05);与模型组相比,利多卡因低剂量与高剂量组心肌组织GSH-Px水平均升高(P<0.05),心肌组织ROS及MDA 水平、血清炎症因子iNOS 及PGE2水平均降低(P<0.05);与利多卡因低剂量组相比,利多卡因高剂量组心肌组织GSH-Px水平均升高(P<0.05),心肌组织ROS及MDA 水平、血清炎症因子iNOS及PGE2水平均降低(P<0.05);与利多卡因高剂量组相比,利多卡因+PI3K 抑制组心肌组织GSH-Px水平均降低(P<0.05),心肌组织ROS及MDA 水平、血清炎症因子iNOS及PGE2水平均升高(P<0.05)。表3。

表3 各组大鼠心肌组织氧化应激因子GSH-Px、ROS、MDA 水平及血清炎症因子iNOS、PGE2 水平
2.4 各组大鼠心肌组织凋亡、自噬与PI3K/AKT通路相关蛋白表达的比较与假手术组相比,模型组心肌组织凋亡蛋白caspase-9及Bax表达水平、自噬蛋白LC3II/LC3I与Beclin-1表达水平显著升高(P<0.05),PI3K/AKT 通路蛋白p-PI3K/PI3K、p-AKT/AKT 水平显著降低(P<0.05);与模型组相比,利多卡因低剂量与高剂量组心肌组织凋亡蛋白caspase-9及Bax 表达水平、自噬蛋白LC3II/LC3I与Beclin-1表达水平均降低(P<0.05),PI3K/AKT通路蛋白p-PI3K/PI3K、p-AKT/AKT 水平均升高(P<0.05);与利多卡因低剂量组相比,利多卡因高剂量组心肌组织凋亡蛋白caspase-9及Bax表达水平、自噬蛋白LC3II/LC3I与Beclin-1表达水平降低(P<0.05),PI3K/AKT 通路蛋白p-PI3K/PI3K、p-AKT/AKT 水平升高(P<0.05);与利多卡因高剂量组相比,利多卡因+PI3K 抑制组心肌组织凋亡蛋白caspase-9 及Bax 表达水平、自噬蛋白LC3II/LC3I与Beclin-1表达水平升高(P<0.05),PI3K/AKT 通 路 蛋 白p-PI3K/PI3K、p-AKT/AKT 水 平降低(P<0.05)。图3、表4。
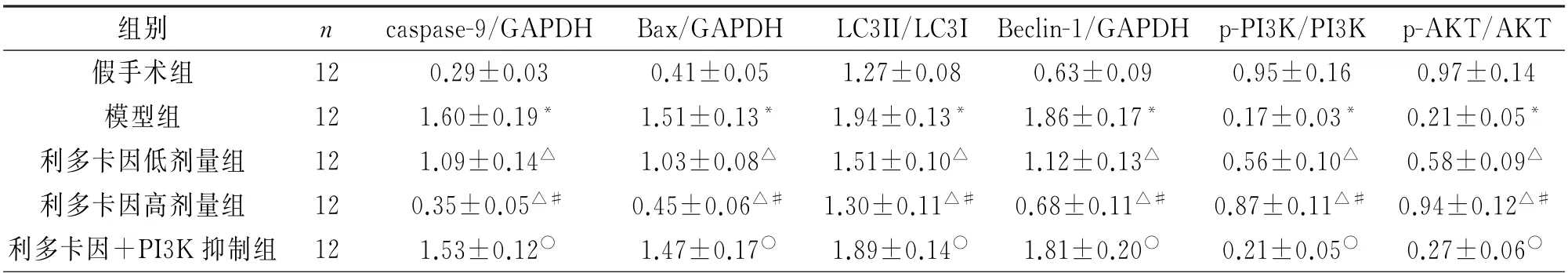
表4 各组大鼠心肌组织凋亡、自噬与PI3K/AKT 通路相关蛋白相对表达水平

图3 免疫印迹检测大鼠心肌组织凋亡、自噬与PI3K/AKT通路相关蛋白表达
3 讨论
心梗在世界范围内发生率不断增加,心律失常作为其诱发的最常见并发症,可使心梗患者心功能严重受损,引发心力衰竭,对人们生命安全的威胁与日俱增,已成为当前急需解决的临床难题[1-2]。本文通过结扎大鼠肺动脉使心肌缺血后再灌注,构建缺血-再灌注心律失常模型,结果显示,心肌缺血-再灌注大鼠炎性细胞因子大量产生,引发严重的炎症及氧化应激反应,造成心肌细胞大量凋亡,导致室颤、室早等心律失常,揭示缺血-再灌注心律失常模型构建成功。
利多卡因被证实参与调控修复心功能,提高心梗患者存活率,改善心律失常症状[12-13]。笔者以不同剂量利多卡因处理缺血-再灌注心律失常大鼠,发现利多卡因可降低心肌组织ROS 及MDA、血清iNOS及PGE2水平,升高心肌组织GSH-Px水平,清除氧自由基,降低炎症因子水平,增强抗氧化能力,抑制心肌细胞凋亡,缓解心肌损伤,减轻室颤、室早等心律失常症状。笔者的结果与既往报道相似,且高剂量利多卡因的疗效更强,进一步证实了利多卡因的抗心律失常作用,但具体的作用机理有待进一步探索。
炎症、氧化应激和自噬是急性心梗后心肌损伤及心律失常的重要病理因素,增强抗氧化活性,减轻心脏炎症及自噬,可显著改善心梗后心功能损伤,降低心律失常发生率[2-3]。PI3K/AKT 是调控组织细胞自噬、凋亡、应激反应及炎症的重要信号,在缺血-再灌注心肌损伤及心律失常的发生、发展过程中发挥着关键作用,促使PI3K/AKT 信号传导,可通过抑制内质网应激和炎症减轻心肌细胞凋亡及心肌过氧化损伤,进而改善心律失常,修复心功能[14-15],并能减弱过度缺血/再灌注引发的心肌自噬,进而减轻心肌损伤[7],笔者推测利多卡因可能通过激活PI3K/AKT 信号,参与改善缺血-再灌注引发的心律失常,结果显示,缺血-再灌注心律失常大鼠心肌组织p-PI3 K/PI3 K及p-AKT/AKT水平显著降低,自噬蛋白LC3II/LC3I与Beclin-1表达水平显著升高,以利多卡因处理后,可增强PI3K/AKT 通路蛋白的磷酸化水平,降低自噬蛋白LC3II/LC3I与Beclin-1表达,表明PI3K/AKT 信号通路通过介导利多卡因对缺血-再灌注心律失常的治疗过程。为进一步证实相关猜想,以PI3K/AKT 信号抑制剂LY294002与利多卡因联合干预缺血-再灌注心律失常大鼠,相比单独使用利多卡因,大鼠心肌损伤及心律失常症状加剧,表明LY294002可减弱利多卡因的抗炎、抗氧化及抑制过度自噬的作用,拮抗利多卡因对心肌损伤的改善功效,逆转其抗心律失常功效,揭示利多卡因可通过激活PI3K/AKT 信号减轻缺血-再灌注损伤引发的心律失常。本实验初步验证利多卡因的调控关系,但PI3K/AKT 信号通路作用广泛,受多种因素调控,是否有其他因子可调控相关通路影响疾病还有待进一步研究,利多卡因作用广泛,是否通过其他通路间接参与调控疾病也有待验证。
猜你喜欢
石家庄学院学报(2021年6期)2021-11-26
云南医药(2021年3期)2021-07-21
服饰导报·鞋世界(2021年4期)2021-05-17
中国药理学通报(2019年5期)2019-01-11
中国医药指南(2017年3期)2017-11-13
中外医疗(2015年5期)2016-01-04
天津医科大学学报(2015年2期)2015-12-22
肿瘤预防与治疗(2015年1期)2015-09-26
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年30期)2015-03-01
