
树形分子内部亲疏水性对其与磷脂膜相互作用影响的动力学研究
2023-05-07 13:23解立强梁盛德朱开礼
兰州理工大学学报 2023年2期
解立强, 梁盛德, 朱开礼
(1. 甘肃民族师范学院 物理与水电工程系, 甘肃 合作 747000; 2. 南京大学 教育部近代声学重点实验室, 江苏 南京 210093; 3. 甘肃民族师范学院 化学与生命科学系, 甘肃 合作 747000)
树形分子是一类具有树枝状结构的纳米分子,具有精确可控的分子结构、内部空腔及大量可修饰的功能团,在医学中有着广泛的应用前景[1-2],如基因传输,癌症诊断等[3-4].截止目前,多种具有不同特性的树形分子被应用于医学特性研究[2,5-8].
树形分子进入人体的第一道障碍是生物膜.为提高其药物转运效率,规避其生物毒性,研究其与生物膜的相互作用变得尤为重要[9].实验上,Mecke等实验发现第三代和第五代PAMAM树形分子能够聚集在磷脂膜上,进而对生物膜产生破坏后果[10-11].他们指出PAMAM树形分子与磷脂膜相互作用会产生树形分子与磷脂分子组成的胶束[10-12].Kelly等[13]实验发现,第六代树形分子与膜相互作用时,会形成磷脂分子包裹树形分子的囊泡结构.Smith等[14]和Mecke等[15]实验发现G5 PAMAM分子能够插入到磷脂膜的内部.Vidal等[16]的实验则支持树形分子是通过内吞作用进入细胞.实验研究还发现,树形分子核心的轻微的改变会对该分子生物特性产生重大影响[2,5].
不同的实验给出了不同的观察结果,预测了不同的作用机制.为确定纳米颗粒与生物膜相互作用机制,学者们开展了大量的理论研究和分子动力学模拟[4].Helfrich[17]、Deserno[18]、Reynwar等[19]和Cao等[20]利用Helfrich的细胞膜连续弹性介质模型,从能量角度出发,研究了纳米颗粒与磷脂膜的作用问题.Gao等[21]和Zhang等[22]采用此模型计算得到25~30 nm的纳米颗粒最容易以内吞的方式进入细胞.应该注意到,利用Helfrich自由能计算时,要求从介观上假定膜的厚度比横向尺度要小很多,进而将膜看成二维的弹性介质薄膜.树形分子的分子尺寸小于10 nm[3-4],因此不能直接使用上述模型进行相应讨论.此时,分子动力学模拟更加适用于研究此类分子与生物膜的相互作用[23].Ginzburg等[24]的动力学模拟结果支持树形分子被磷脂膜包裹为一个球状结构.Lee和Larson的粗粒化模拟表明树形分子能够穿过膜,不会形成囊泡结构[25-27].这些相互矛盾的结果,使得树形分子与膜相互作用机制更加扑朔迷离.前期工作表明,只有代数不小于5的PAMAM树形分子能被磷脂膜包裹,形成囊泡结构[28].模拟结果与部分的理论模型一致,但也与部分的模型相悖.树形分子与膜之间相互作用的分子机制仍需进一步阐释.
本文中,采用MARTINI粗粒化分子动力学模拟的方法[29-30],重点研究了树形分子内部的亲疏水性对其与磷脂膜相互作用的影响.研究发现,生物膜与树形分子间的静电相互作用能够将树形分子锚定到膜表面,而树形分子内部的亲疏水性对其与磷脂膜的作用具有很强的调制性.具有强疏水核心的树形分子能够将磷脂分子拉出膜,形成胶束结构,进而造成膜两侧的不对称及膜局部的缺陷,从而利于树形分子的穿膜运动.模拟研究还发现,模拟系统尺寸的大小,即膜的大小,对模拟结果有着显著影响.研究结果有利于解释目前树形分子与生物膜相互作用的不同机制,对树形分子理论研究、合成及医学应用具有重要的理论指导意义.
1 模型与方法
本文以第五代(G5)的树形分子为研究对象,采用MARTINI粗粒化力场2.1版本进行分子动力学模拟[29-30].粗粒化的G5 PAMAM树形分子[25]和DMPC磷脂分子[31]拓扑结构可以直接从Marrink小组的网站下载.
在MARTINI力场中,G5 PAMAM(聚酰胺胺)树形分子的节点和分支分别用N0和Nda珠子表示[28].为研究具有疏水核心的树形分子,在G5 PAMAM树形分子的基础上,将其分支上的珠子替换成了力场中具有强疏水特性的C1珠子,以构造具有强疏水核心特性的树形分子.为方便讨论,G5 PAMAM分子用M1表示,新构造的分子用M2表示.图1给出了代数为1的两种树形分子的拓扑结构,更高代数的树形分子可以通过在端点叠加单体实现.在结构上,M2更接近PPI树形分子[32].在pH=7时,这两种分子端点的氨基全部被质子化[3-4],在该力场中用Qd珠子表示,且带了1个单位的正电荷.这样,一个G5树形分子具有128个单位的正电荷.将树形分子置于水溶液中,对系统进行电中和,分别进行动力学平衡模拟,研究其分子结构特性.

图1 第一代树形分子结构示意图
模拟研究表明,带正电的树形分子能够吸引双层膜上负电的磷脂分子,促进磷脂分子向树形分子游动,形成负电磷脂分子的畴结构[28].本文直接使用负电的DMPG(二肉豆蔻酰磷脂酰甘油)分子组成的磷脂膜作为研究对象.128个DMPG磷脂分子组成的双层膜可以由Marrik小组网站上的DMPC双层膜调整而来.将DMPC分子头部的Qd珠子替换成了P4珠子,带电量设为0,即得到一个由128个DMPG分子组成的负电磷脂膜.在系统中加入128个NA+进行电中和,温度设定为320 K,对系统进行平衡200 ns,得到一个稳定的DMPG双层膜.将该双层膜在XY平面上沿着X和Y方向将系统各扩展2倍或者6倍,就得到了含有512和4 608个磷脂分子的DMPG磷脂膜,对两个系统分别进行200 ns的平衡.
平衡好后,在Z轴方向上,将平衡好的树形分子放置到离膜中心3~4 nm的地方,对树形分子和磷脂膜分别用CL-和NA+中和.将树形分子和磷脂膜分子进行弹性约束,对系统中的水分子和离子进行平衡运算200 ns.解除约束,考察树形分子与生物膜相互作用的动力学过程.对应的模拟系统见表1.
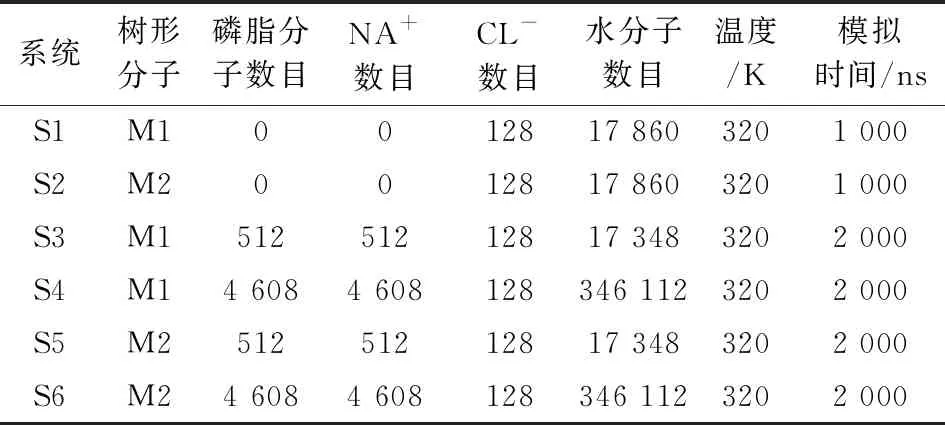
表1 模拟系统的参数
所有模拟采用MARTINI力场2.1版本标准动力学参数并在NPT系综中进行.压强设定为1个标准大气压,温度设定为320 K,系统压强/温度采用Berendsen热浴[33].范德华相互作用采用Lennard-Jones(LJ)作用势,截断距离为1.2 nm(cut-off),且LJ势在0.9和1.2 nm之间平滑衰减为0.离子间的静电相互作用截断距离为1.2 nm,静电势能从0至1.2 nm平滑衰减为0.
水的相对介电常量在该力场中被设为15,时间积分步长设置为20 fs.模拟采用GROMACS5.0动力学模拟软件[34],分子动力学轨迹的可视化利用VMD软件实现[35].在Martini力场中,模拟时间对应全原子模拟时间的4~8倍[29-30],正文描述中使用的是模拟时间.
2 结果与讨论
2.1 具有不同疏水核心的树形分子的构像
利用S1和S2系统,研究了水溶液中M1与M2分子的构象.如图2所示,经过1 000 ns,两种分子的回旋半径都趋于稳定.M1分子的回旋半径为2.52 nm,M2为2.41 nm.可以看到,虽然两者的亲疏水核心相差很大,但两者之间的差距很小.其中M1分子的结果与之前的模拟结果一致[28].而M2分子,在结构上与PPI的分子类似,因此,将结果与PPI分子的相关结果进行比较.可以发现,M2分子的回旋半径稍微大于PPI分子全原子模拟(AAMD)的结果[32],主要原因为M2分子是直接由M1分子改造而来,没有对键长和键角等进行额外修饰,因此M2分子仅仅代表具有强疏水核心的树形分子.

图2 两种树形分子回旋半径与时间的关系
图3给出了水分子相对于树形分子中心的径向分布函数.对于两种树形分子,在0.5 nm处都有一个很高的峰,表明在树形分子的中心处水分子都有一个较高的分布.这主要是因为在粗粒化模型中,M1和M2分子中心是一样的.在分子中心与端点之间,水分子相对于M1树形分子核心的分布明显要低于M2分子,而在端点区域,结论相反.通过分析树形分子的结构特点和构像可知(图1),产生该现象的原因主要是M2分子的内部单体比M1分子更具有疏水性,在水溶液中,M2分子的单体之间呈现出紧密的靠拢趋势,而不是简单的向核心塌缩.这样,在树形分子中可有更大的暴露空间,使得水分子有更大的概率进入M2树形分子内部空腔.这也解释了为什么两种树形分子的回旋半径会如此接近.

图3 水分子相对于树形分子中心的径向分布函数
2.2 M1树形分子与磷脂膜的相互作用
利用S3系统来考察M1分子与512个DMPG分子组成的双层膜间的相互作用.图4给出了系统S3和S4的动力学过程.图4a~4d分别对应系统S3和S4的初始和末了构像.M1树形分子被表示为绿色.磷脂分子的碳链表示成青色,带电头部表示成橙色.为清晰期间,水分子和离子未显示,下文中采取相同的颜色标识.

图4 树形分子M1与磷脂膜的相互作用
在S3系统中,经过2 000 ns的动力学过程,发现树形分子会紧紧的粘附到膜的上表面,观察不到膜有明显的形貌变化.虽然在模拟中使用了周期性的边界条件来模拟无限大的生物膜,但由于树形分子镜像的影响,抑制了膜的涨落.而膜的涨落对于纳米粒子与膜的相互作用是一个重要影响因素[36].为此,需要考虑树形分子与大尺寸膜间的相互作用.S4系统对应于M1分子与一个由4 608个DMPG分子组成的磷脂膜的相互作用.从图4可以看到,经过2 000 ns的动力学过程,在S4系统中,双层膜有一个明显的包裹树形分子M1的趋势.比起1∶1 DMPC/DMPG组分的磷脂膜,这种包裹趋势更加明显[28].能量分析指出,这种膜弯曲的驱动力是树形分子端点和磷脂分子带电头部的静电力驱动的[28].
图5给出了系统S3和S4中树形分子与磷脂膜之间距离随着时间的变化关系.通过对比可以看到,动力学过程中,膜的尺寸越大,膜的涨落越大,树形分子与磷脂膜中心的距离涨落也越大.稳定后,膜越大,对树形分子包裹的程度越大.研究指出,模拟的结果依赖于模拟系统的大小[37].
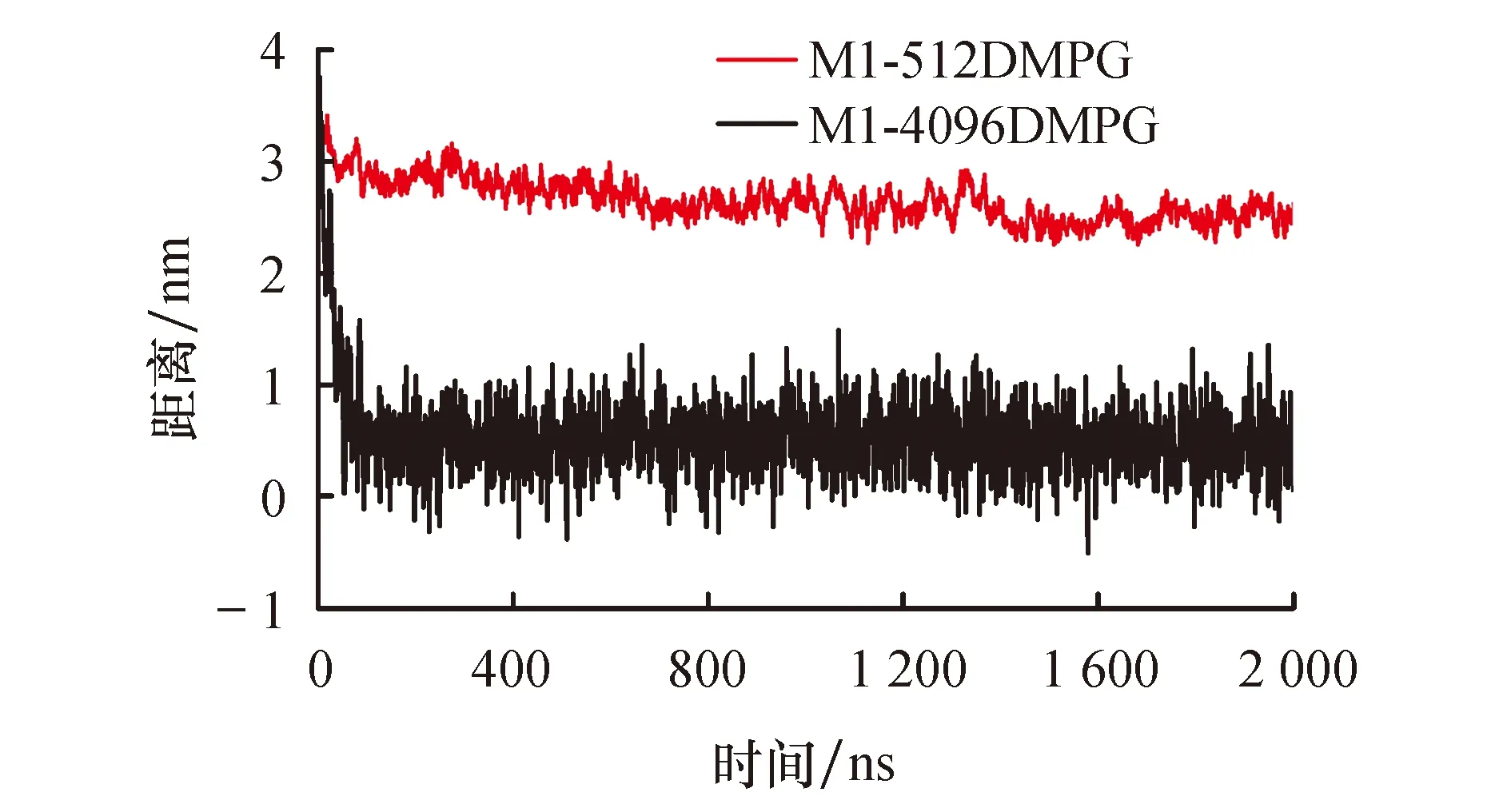
图5 M1树形分子与磷脂分子质心间的距离(z轴)与时间关系
上述模拟表明,对于负电膜,M1树形分子不能穿膜,这与AAMD模拟一致.AAMD指出,对于PAMAM树形分子,细胞膜提供了一个很大能垒,使得PAMAM树形分子只能粘附到膜的表面[38-39].从这个角度出发,M1树形分子更倾向于采取吸附介导的内吞作用进入细胞[16,28].
2.3 M2树形分子与磷脂膜的相互作用
图6给出了S5系统的动力学过程,对应M2分子与512个DMPG分子组成的双层膜间的相互作用.在图6中,M2树形分子被表示为蓝色.模拟同样发现M2树形分子同样会很快粘附到膜上表面.800 ns时,有DMPG分子陆续被拉出双层膜(在图6中,该类磷脂分子用红色表示).在拉出的过程中,磷脂分子的碳链保持在树形分子内部,负电的头部始终与树形分子带正电的端点相接触.脱离双层膜的分子在树形分子中不停的游走.经过2 000 ns的动力学过程,共有5个分子脱离双层膜.实验和模拟都证实,磷脂分子可以进入树形分子空腔[40-42].但要注意到,他们是利用树形分子与磷脂分子的自组装方法得到的,而本文第一次在分子动力学模拟中观察到磷脂分子被树形分子从膜中拉出并进入树形分子空腔的现象.

图6 树形分子M2与512个磷脂分子构成的双层膜的相互作用(S5)
与树形分子接触的膜的上表面由于被拉出磷脂分子,会造成膜的上下两层的不对称.磷脂膜上层由于缺少磷脂而导致膜的弯曲,进而促进树形分子与膜的进一步相互作用[28,43].当所采用的膜较大时,上层磷脂分子的游离,会诱引膜自发曲率的产生[17].
图7给出了M2分子与4 608个磷脂分子组成的磷脂膜相互作用的动力学过程.模拟发现M2分子在膜涨落的辅助下会更快的被磷脂膜包裹(t=20 ns).随着树形分子一个疏水单体与磷脂膜疏水部分的接触,磷脂分子逐渐被拉出膜(t=200 ns).在t=450 ns时,磷脂分子与树形分子形成胶束结构.这种结构是第一次在模拟树形分子与膜的相互作用中观察到.Lind等[44]和Evans等[45]的实验发现树形分子和磷脂分子能够在磷脂膜上形成球状胶束结构,模拟结果与实验观察一致.Lee等[42]的动力学模拟中,这种胶束结构的实现只能通过自组装实现.在相同的时间内,在S5系统中没有观察到这种胶束结构.

图7 树形分子M2与由4 608个磷脂分子构成的双层膜的相互作用
500 ns时,树形分子的一个单体开始穿透膜,并与膜下层的磷脂分子头部接触.同时观察到在与树形分子接触的磷脂膜上形成了局域的缺陷,使得水分子能够进入膜的内部,见图7(t=500 ns).在接下来的100 ns内,有多个树形分子端点陆续穿过膜.胶束结构开始与膜逐渐融合,形成M2分子镶嵌在膜中的凸起结构,并持续到模拟最后.由图8可知,在分子动力学最后,这种凸起结构位于膜中心的上部.对模拟过程进行分析可以观察到,对于尺度比较大的膜,磷脂分子能够更快、更多的被拉出膜,进而促进M2分子进入磷脂膜.比较图3和6,可以看到具有疏水核心的M2分子对膜的扰动更大.

图8 M2树形分子与磷脂分子质心间的距离(z轴)与时间关系
实验上观察到树形分子能够将磷脂分子从膜上带走,进而对膜产生破坏作用[10,12-13,15].在本模拟中没有观察到胶束结构脱离膜.如果能够脱离膜,则可以解释实验上观察到膜破裂的情况.但应注意到,本文只是模拟了一个树形分子与膜的相互作用.如果有多个的树形分子与膜同时相互作用,会有更多的磷脂分子脱离膜,进而导致膜较大面积的收缩,甚至破裂.膜的破裂会有助于胶束结构脱离膜及膜上空洞的产生[28,46].
模拟最后得到的凸起结构中心位于膜中心的上部(见图7和图8),这能够解释为什么实验上人们观察到带电的树形分子能够将支撑膜上的磷脂分子脱离,而不是粘附到基底上[10,12-13,15].
2.4 讨论
由模拟结果可以看出,磷脂分子被拉出双层膜不能简单归于树形分子带电,因为带同样电荷的M1分子不能拉出磷脂分子(S3和S4系统).Lee等[25]指出,在MARTINI力场中,只有结合静电的长程作用(PME方法)才能观察到M1分子直接穿过膜,尽管他们指出,使用PME与普通的无极性水模型,对双层膜物理性质的影响较小,但应注意到当水分子采用非极性珠子时,水的介电常数被设置为15来代表水的静电屏蔽作用.这导致Lee等[25]的结果与全原子模拟是矛盾的.全原子模拟没有观察到PAMAM树形分子的穿膜运动,能量分析指出该运动会受到磷脂双层膜提供的一个很高的能垒[38-39].本文与全原子模拟一致,静电作用仅仅使得树形分子M1能够粘附到表面上,而核心的疏水性在树形分子M2的穿膜过程中起到了至关重要的作用[43,47].因此,本文指出,树形分子能够穿过磷脂膜仅仅是由于静电的相互作用是不合理的.模拟中将介电常数设定为一个固定的常数,将会导致极性分子在非极性环境下的作用被低估.Yesylevskyy等[48]后期开发了充分显示水极性的力场.在该力场中,由于考虑了水的极性,可以使用PME方法来计算静电的长程作用.在小系统中(S3和S5),两种力场所得结果是一致的.
研究发现膜的尺寸对研究对象的影响不应该忽略.系统越大,消耗更多的计算量,但是在膜的涨落辅助下,达到同一动力学状态,却需要较少的动力学时间.有些现象甚至在尺寸小的系统中是观察不到的.为了使得模拟结果更加科学可靠,在研究纳米粒子,特别是对于树形分子这种大分子与膜的相互作用时,膜的尺度应当尽可能的大.
树形分子由于带正电,因此更喜欢负电的磷脂膜[49-50].之前的研究表明,树形分子会首先吸引负电的磷脂分子到树形分子底部,形成畴结构,继而引起膜的弯曲[28].本文采用了一个全部是负电的单组分磷脂膜,在一定程度上加快了树形分子与膜的粘附过程.考虑到实验的复杂性,树形分子对膜的破坏应该需要更长的时间[10,12-13,15].
在本文的模拟中,仅仅考察了一个树形分子与磷脂双层膜的动力学过程.可以推测的是,当多个树形分子同时与膜相互作用时,会对应着双层膜较大面积的收缩.在实验上,这种膜面积的收缩,会在囊泡表面上产生膜缺陷,导致囊泡内部物质的泄露或者囊泡的破裂[51].因此,随着计算能力的提升,考虑到实验的复杂性[50],树形分子的毒性对于浓度的依赖及分子潜在的协同作用要考虑,多个分子同时或者陆续与膜或囊泡的相互作用值得被研究[43,52-55].
对于本文中的M2分子,代表了一类具有强疏水核心的树形分子,并没有针对特定的分子.在医学应用中,人们已经合成了具有不同特性的树形分子[6-7],这些树形分子与膜的相互作用机制仍需继续开展相关理论与模拟研究.本文的结果可为后期的理论与实验研究提供新的启示,即通过调节树形分子的内部属性来调节其与生物膜的相互作用.
3 结论
本文采用MARTINI粗粒化分子动力学模拟的方法,重点研究了树形分子内部的亲疏水性对其与生物膜相互作用的影响.发现树形分子与磷脂膜的相互作用对树形分子内部的亲疏水性具有极强的依赖性.内部核心疏水性强的树形分子能够粘附到磷脂膜表面,并将磷脂分子从膜中拉出,形成树形分子与磷脂分子组成的胶束.树形分子对膜形成较大扰动的同时,能够穿过膜,造成膜的穿孔.内部核心具有弱疏水性的树形分子仅仅粘附到磷脂膜的表面,形成磷脂膜包裹树形分子结构.研究表明,模拟系统的尺寸的大小,即膜的大小,对模拟结果有着显著影响.不同的模拟尺寸在相同的时间里给出不同的观察结果.本文的研究结果有利于解释目前树形分子与生物膜相互作用的不同机制,对树形分子的合成及医学应用具有重要指导意义.
致谢:本文得到甘肃民族师范学院校长科研基金(GSNUXM21-7)和甘肃省高等学校青年博士支持项目的资助,在此表示感谢.
猜你喜欢
河北果树(2022年1期)2022-02-16
现代临床医学(2021年5期)2021-11-02
昆明医科大学学报(2021年4期)2021-07-23
烟台果树(2021年2期)2021-07-21
中成药(2019年12期)2020-01-04
中成药(2018年7期)2018-08-04
现代园艺(2017年19期)2018-01-19
现代园艺(2017年13期)2018-01-19
中成药(2017年12期)2018-01-19
中成药(2017年5期)2017-06-13
