
鞘氨醇激酶1(Sphk1)抑制剂在肝纤维化大鼠模型中的作用机制
2023-04-29 16:54:04王燕马丽肖雄雷冬梅赵奉茹郭力源王晓忠
临床肝胆病杂志 2023年8期
王燕 马丽 肖雄 雷冬梅 赵奉茹 郭力源 王晓忠


摘要:目的探討鞘氨醇激酶1(SphK1)抑制剂在不同肝纤维化大鼠模型中的治疗作用。方法170只SD大鼠随机分为4组:正常组(30只)、HFE组(给予高脂乳剂,45只)、CCl4组(CCl4诱导,45只)、CCl4+HFE组(HFE联合CCl4,50只),经病理及实验室检查证实造模成功后,分别给予SphK1抑制剂PF-543,同组别以生理盐水作为对照,分别在第1、7、14天取肝组织进行Masson染色,比较肝纤维化面积占比、透射电镜下观察自噬小体形成情况、Real-time PCR检测肝纤维化及自噬相关标志物mRNA表达水平。计量资料多组间比较采用单因素方差分析,进一步两两比较采用LSD检验。相关性分析采用Spearman相关分析。结果与正常组(0.57±0.13)相比,CCl4组(6.93±5.81)和HFE+CCl4组(10.89±2.67)肝纤维化面积占比均升高(P值均<0.01)。PF-543干预第7天可显著降低CCl4组及HFE+CCl4组纤维化病变面积(P值均<0.01)。与正常组相比,HFE组、CCl4组和HFE+CCl4组的SphK1表达水平均明显降低(P值均<0.01),HFE+CCl4组的α平滑肌肌动蛋白、转化生长因子-β、α1-Ⅰ型胶原mRNA表达水平均明显升高(P值均<0.01),干预后各指标水平无明显变化(P值均>0.05)。Atg5、Atg12、Becn1的表达水平与肝纤维化病变面积均呈负相关(r分别为-0.715、-0.640、-0.632,P值均<0.01);SphK1的表达水平与Atg5、Atg12、Becn1、Map1lc3a 的mRNA表达均呈正相关(r分别为0.603、0.561、0.510、0.498,P值均<0.01)。电镜示:CCl4组肝组织轻微水肿,细胞器丰富,轻度肿胀,以粗面内质网扩张为主。该视野可见9个自噬溶酶体结构。在PF-543 干预第7天后,未见典型自噬结构。HFE+CCl4组肝组织轻度水肿,结构不清晰,粗面内质网数量丰富,明显扩张,表面附着核糖体颗粒较少。该视野可见6个自噬溶酶体结构。在PF-543 干预第7天后,该视野未见典型自噬结构。结论PF-543对自噬有显著抑制作用,并同时伴有纤维化面积的减少。提示靶向SphK1,可影响到肝脏的自噬水平,并进而缓解两种肝纤维化模型的纤维化状态。
关键词:肝纤维化; 鞘氨醇激酶1; 自噬; 模型, 动物
基金项目:国家自然科学基金(81860808)
Mechanism of action of sphingosine kinase 1 inhibitor in a rat model of liver fibrosis
WANG Yan MA Li XIAO Xiong LEI Dongmei ZHAO Fengru GUO Liyuan WANG Xiaozhong (1. The Fifth Peoples Hospital Affiliated to Chengdu University of Traditional Chinese Medicine, Chengdu 611130, China; 2. Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830000, China; 3. Traditional Chinese Medicine Hospital of Xinjiang Uygur Autonomous Region, Urumqi 830000, China)
Corresponding author: WANG Yan, wy751105@126.com (ORCID:0000-0001-9730-8279)
Abstract:ObjectiveTo investigate the therapeutic effect of sphingosine kinase 1 (SphK1) inhibitor in different liver fibrosis models. MethodsA total of 170 Sprague-Dawley rats were randomly divided into normal group (30 rats), HFE group (45 rats given high-fat emulsion), CCl4group (45 rats induced by CCl4), and CCl4+HFE group (50 rats given HFE and CCl4). After successful modeling confirmed by pathology and laboratory examination, the SphK1 inhibitor PF-543 was given, and the rats in the same group were given normal saline as control. Liver tissue samples were collected on days 1, 7, and 14 for Masson staining; the percentage of liver fibrosis area was compared; the formation of autophagosome was observed under a transmission electron microscope; real-time PCR was used to measure the mRNA expression levels of markers associated with liver fibrosis and autophagy. A one-way analysis of variance was used for comparison between multiple groups, and the LSD method was used for further comparison between two groups; a Spearman correlation analysis was also performed. ResultsCompared with the normal group, the CCl4group and the HFE+CCl4group had a significant increase in the percentage of liver fibrosis area (6.93±5.81/10.89±2.67 vs 0.57±0.13, both P<0.01), the CCl4group and the HFE+CCl4group had a significant reduction in the percentage of liver fibrosis area on day 7 (both P<0.01). Compared with the normal group, the HFE group, the CCl4group, and the HFE+CCl4group had a significant reduction in the expression level of SphK1 (all P<0.01), and the HFE+CCl4group had significant increases in the mRNA expression levels of alpha-smooth muscle actin, transforming growth factor-β, and collagen type I alpha 1 (all P<0.01), while there were no significant changes in these indices after intervention (all P>0.05). The expression levels of Atg5, Atg12, and Becn1 were negatively correlated with the area of liver fibrosis (r=-0.715, -0.640, and -0.632, all P<0.01), and the expression level of SphK1 was positively correlated with the mRNA expression of Atg5, Atg12, Becn1, and Map1lc3a (r=0.603, 0.561, 0.510, and 0.498, all P<0.01). Electron microscopy showed that the CCL4group had slight edema, abundant organelles, and mild swelling in liver tissue, mainly the expansion of rough endoplasmic reticulum, and nine autolysosome (ASS) structures were seen in this field, while no typical ASS structure was observed on day 7 of PF-543 intervention. The HFE+CCL4group had mild edema, unclear structure, and abundant rough endoplasmic reticulum with marked expansion and few ribosome particles attached to its surface in liver tissue, and 6 ASS structures were seen in this field, while no typical ASS structure was observed on day 7 of PF-543 intervention. ConclusionPF-543 significantly inhibits autophagy and is associated with a reduction in fibrosis area. It is suggested that targeting SphK1 can affect the level of liver autophagy, thereby alleviating the state of liver fibrosis in two liver fibrosis models.
Key words:Hepatic Fibrosis; Sphingosine Kinase 1; Autophagy; Models, Animal
Research funding:National Natural Science Foundtion of China(81860808)
全球有15亿人受到多种形式慢性肝病的影响[1]。肝纤维化被认为是肝脏结构恶化的节点。近年来,由于肝炎、非酒精性脂肪性肝炎等所致肝硬化的发病率及其相关病死率急剧增加[2]。不同病因所致肝硬化潜在的发病机制和在疾病进展、治疗效应上的差别是未知的。迫切需求探索可减轻肝纤维化的新细胞与分子途径。
鞘氨醇激酶(SphK)蛋白是一个激酶家族,负责鞘氨醇的磷酸化和鞘氨醇-1-磷酸(S1P)的产生,调节不同的生物事件,如细胞增殖、凋亡、迁移和血管生成[3]。S1P是一种具有广泛生物活性的磷脂酶,由SphK1催化而来。SphK1所产生鞘氨醇-1-磷脂,已被证明可调节肥胖人类和小鼠的肝脏脂肪含量、脂肪组织炎症及胰岛素抵抗[4]。有报道[5]SphK1抑制剂可调节巨噬细胞分泌肿瘤坏死因子α,可改善小鼠暴发性肝衰竭。SphK1缺乏可显著提高小鼠对对乙酰氨基酚诱导肝损伤的耐受能力[6]。SphK1在高饱和脂肪喂养诱导的小鼠非酒精性脂肪性肝炎模型中介导肝脏炎症,并启动肝细胞促炎信号传导。SphK1-/-可保护肝脏免受胰岛素抵抗[7],研究还表明,对代谢功能障碍的其他参数,包括血浆游离脂肪酸,SphK1没有保护作用。相反,另有研究[8]证实,在非酒精性脂肪性肝病肝脏缺血再灌注损伤(I/R)的小鼠模型中敲低SphK1可引起脂毒性的神经酰胺生成增加,加重肝细胞氧化应激,引起细胞凋亡与坏死,进而加重肝脏I/R损伤。这表明在肥胖背景下,SphK1在代谢异常中的作用是多样化的。新近研究[9]发现,SphK1能够通过诱导自噬参与多种疾病的发生发展。在四氯化碳(CCl4)诱导的小鼠肝纤维化模型中,检测到肝组织中的微管相关蛋白1轻链3-Ⅱ表达增加,说明自噬水平在肝纤维化中明显增加。饥饿状态下,SphK1激活自噬,抑制了 MCF-7细胞的死亡,保证了细胞存活[10]。
在本研究中,设计了肝纤维化和脂肪肝肝纤维化两种大鼠模型,两者造模时间均为12周。运用 SphK1抑制剂PF-543干预,比较两种不同纤维化模型对SphK1抑制剂反应是否相同,探讨SphK1在不同病因所致肝纤维化中的作用及机制。
1材料与方法
1.1肝組织与动物SD大鼠购自新疆医科大学实验动物中心[实验动物生产许可证号:SCXK(新)2018-0003,实验动物使用许可证号:SYXK(新)2018-0003],雄性,年龄5~6周龄,无特殊病原级,平均体质量176.7 g。SD大鼠共170只,随机分为4组:正常组、高脂乳剂组(high fat emulsion,HFE)、四氯化碳组(CCl4)、高脂乳剂+四氯化碳组(HFE+CCl4),其中Normal组30只、HFE组45只、CCl4组45只、HFE+CCl4组50只。
正常组每天灌胃给水+每周三、日皮下注射生理盐水;HFE组每天10 mL/kg灌胃高脂乳剂;CCl4组每周三、日按3 mL/kg皮下注射CCl4; HFE+CCl4组每天按10 mL/kg灌胃高脂乳剂+每周三、日按3 mL/kg皮下注射CCl4,连续12周。分别于造模第4、8、12周各组处死指定数量大鼠。造模13周结束后二次分组,各组分别给予PF-543(SphK1抑制剂)及生理盐水,按照剂量0.5 mg/kg、浓度0.2 mg/mL、体积2.5 mL/kg 腹腔注射PF-543及等体积生理盐水。各组分别于注射后第1、7、14天(D1、D7、D14)处死动物,取肝组织,部分置于2.5%戊二醛中固定,进行电镜观察;部分肝组织置于4%多聚甲醛中固定,用于光镜观察、Masson染色。
1.2HE染色常规烤片及脱蜡处理,酒精水化后进行染色,苏木精复染3 min,盐酸酒精分化1~2 s后置于自来水中终止分化。脱水后伊红复染:0.5%伊红乙醇液对切片进行对比染色,染色1 min后放入95%乙醇中清洗,清洗掉多余的红色后放入无水乙醇中浸泡5 min。二甲苯Ⅰ、Ⅱ 各5 min进行透明。中性树胶封片。
1.3Masson染色4%多聚甲醛中固定24 h的组织标本经修剪、脱水、包埋、切片、染色、封片,最后镜检合格的样片。使用PANNORAMIC全景切片扫描仪将组织切片上机扫描成像,形成文件夹,该文件夹包含了组织切片上所有的组织信息。文件夹用CaseViewer 2.4软件打开后可以1~400倍任意倍数放大后进行观察。使用Halo v3.0.311.314分析软件中Indica Labs-Area Quantification v2.1.3模块分别定量每张切片的目的区域,测量出组织面积、阳性面积并得出阳性面积占比。
1.4透射电镜下观察自噬体新鲜组织确定取材部位,组织体积一般不超过1 mm×1 mm×1 mm,迅速投入电镜固定液4 ℃固定2~4 h。0.1 mmol/L磷酸缓冲液PB漂洗3次,每次15 min。后固定:1%锇酸·0.1 mmol/L磷酸缓冲液PB室温(20 ℃)固定2 h。0.1 mmol/L磷酸缓冲液PB漂洗3次,每次15 min。脱水:组织依次入50%-70%-80%-90%-95%-100%-100%酒精-100%丙酮-100%丙酮上行脱水,每次15 min。渗透:丙酮︰812包埋剂=1∶1, 2~4 h; 丙酮∶812包埋剂=2∶1,渗透过夜;纯812包埋剂5~8 h,将纯812包埋剂倒入包埋板,将样品插入包埋板后37 ℃烤箱过夜。包埋:60 ℃烤箱聚合48 h。切片:超薄切片机切片60~80 nm超薄切片。染色:铀铅双染色(2%醋酸铀饱和酒精溶液,枸橼酸铅,各染色15 min),切片室温干燥过夜。透射电子显微镜下观察,采集图像分析。
1.5Real-time PCR检测mRNA表达水平采用HieffTM qPCR SYBR Green Master Mix (Low Rox Plus)(Yeasen)和特异性引物进行扩增,扩增条件如下:预变性95 ℃ 5 min,变性95 ℃ 10 s,退火延伸60 ℃ 30 s,扩增40个cycle,熔解曲线采集95 ℃ 15 s,60 ℃ 60 s,95 ℃ 15 s。采用2-ΔΔCT方法测定基因相对表达量,并针对GAPDH进行归一化处理,引物序列见表1。
1.6实验试剂高脂乳剂(20%猪油200 g、10%胆固醇100 g、0.5%丙基硫氧嘧啶5 g、4%吐温-80 40 mL、1%猪胆酸盐10 g、加蒸馏水至1 000 mL)、胆固醇(批号:180110,上海蓝季生物科技发展有限公司)、丙基硫氧嘧啶(批号:20201011,上海蓝季生物科技发展有限公司)、吐温(批号:20200820,天津市北联精细化学品开发有限公司)、橄榄油[批号:B20200612X,嘉里粮油(天津)有限公司]、10%中性福尔马林固定液(批号:0603A20,0225A21,安徽雷根生物技术有限公司)、2.5%戊二醛固定液(批号:0122A21,安徽雷根生物技术有限公司),PF-543(99.34%,批号:S717703,美国Selleckchen公司)、Dimethyl sulfoxide
(批号:1129E0312,北京索莱宝科技有限公司)。荧光定量PCR实验试剂:One-Step gDNA Removal and cDNA(批号:AT311-03,Transgene);HieffTM qPCR SYBR Green Master Mix(批号:11202ES08,Yeasen)
1.7统计学方法采用GraphPad 8.0软件对数据进行分析。计量资料以x±s表示,多组间比较采用单因素方差分析,进一步两两比较采用LSD检验。相关性分析采用Spearman相关分析。P<0.05为差异有统计学意义。2结果
2.1各组动物造模后肝组织病理学观察图1显示,正常组肝细胞索排列有序,肝细胞未见变性、坏死,肝窦未见淤血。HFE组可见明显肝细胞空泡变性(++++)。CCl4组可见肝细胞空泡变性(++++),间质结缔组织增生,炎症细胞浸润,符合肝纤维化表现;HFE+CCl4组肝细胞空泡变性,假小叶形成,间质炎症细胞浸润,符合肝硬化表現。
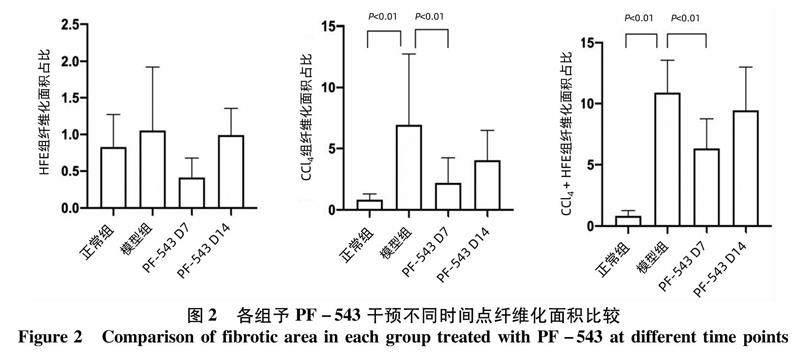
2.2各组动物第1、7、14天给予PF-543对肝纤维化面积占比的影响 两种肝纤维化模型组肝纤维化面积(CCl4组:6.93±5.81,HFE+CCl4组:10.89±2.67)均较正常组(0.57±0.13)升高(P值均<0.01);各组经PF-543干预第7天,CCl4组(2.16±2.07)、HFE+CCl4组(6.32±2.42)肝纤维化面积均有改善(P值均<0.01);在干预第14天时各组肝纤维化面积较第7天略有升高,与各自模型组相比差异无统计学意义(P值均>0.05)。HFE组的肝纤维化面积在干预前后无明显变化(P值均>0.05)(图2)。
如图3所示,正常组肝组织经Masson染色,未见明显的胶原纤维增生。HFE组肝组织脂肪变性明显,大量的肝细胞胞质内可见数量不等的圆形空泡。CCl4组、HFE+CCl4组肝小叶结构破坏,广泛胶原纤维明显增生,染色呈蓝色;增生的胶原纤维将肝细胞分割包绕成大小不等的圆形或椭圆形的肝细胞团,广泛可见假小叶形成;肝组织脂肪变性明显,大量的肝细胞胞质内可见数量不等、体积大小不一的圆形空泡。
CCl4组给予PF-543干预第1天,肝小叶结构破坏,广泛胶原纤维明显增生;增生的胶原纤维将肝细胞分割包绕成大小不等的圆形或椭圆形的肝细胞团,广泛可见假小叶形成。干预第7天,可见几处胶原纤维轻度增生;较多的血管之间胶原纤维相互延伸、连接,形成纤维桥接;肝组织脂肪变性明显,大量的肝细胞胞质内可见数量不等、体积大小不一的圆形空泡。干预第14天,组织边缘局部可见胶原纤维增生;肝组织脂肪变性明显,大量的肝细胞胞质内可见数量不等、体积大小不一的圆形空泡,相较于第1天,肝脏病理可见明显改善。
HFE+CCl4组干预第1天,肝小叶结构破坏,广泛胶原纤维明显增生;增生的胶原纤维将肝细胞分割包绕成大小不等的圆形或椭圆形的肝细胞团,广泛可见假小叶形成;肝组织脂肪变性明显,大量的肝细胞胞质内可见数量不等、体积大小不一的圆形空泡。干预第7、14天,肝脏病理未见明显改善。
2.3PF-543对SphK1及肝纤维化相关基因表达的影响Real-time PCR结果显示,与正常组相比,HFE组、CCl4组和HFE+CCl4组的SphK1表达水平均明显降低(P值均<0.01)。经PF-543干预7天后,HFE组与CCl4组SphK1表达水平仍低于正常组(P值均<0.01)。与正常组相比,CCl4组α平滑肌肌动蛋白(α-SMA)表达水平升高(P<0.01),干预后其表达水平仍高于正常组(P<0.05)。TGF-β表达水平随着纤维化程度加重而逐步增高,其中HFE+CCl4组表达水平最高,与正常组相比差异有统计学意义(P<0.01)。干预前后TGF-β表达水平无明显变化(P>0.05)。Col1a1表达水平随着纤维化程度加重而逐步增高,CCl4组、HFE+CCl4组与正常组相比差异均有统计学意义(P值均<0.01),干预前后其表达水平无明显变化。Fn1表达水平随着纤维化程度加重而逐步增高(P值均<0.01),经PF-543干预后,2个纤维化组表达水平仍低于正常组(P<0.01)(图4)。
2.4自噬相关基因表达与肝纤维化面积的相关性分析如图5所示,Atg5、Atg12、Becn1的表达与Masson阳性面积均呈负相关(P值均<0.01)。Map1lc3a的表达与Masson面积无相关性(P>0.05)。SphK1表达水平与Atg5、Atg12、Becn1、Map1lc3a 的mRNA表达均呈正相關(P值均<0.01)。
2.5SphK1抑制剂PF-543对纤维化肝组织自噬结构的影响电镜下观察正常组可见少量脂滴微自噬结构,未见典型自噬结构。HFE组脂滴局部分布,少量轻度融合,脂滴微自噬结构少量可见。初级溶酶体局部分布,未见典型自噬结构。正常组与HFE组给予PF-543后无明显变化(视野无或可见1个自噬溶酶体)。CCl4组肝组织轻微水肿,膜完整、结构清晰,胞内基质电子密度均匀,细胞器丰富,均匀分布,轻度肿胀,以粗面内质网扩张为主。该视野可见9个自噬溶酶体结构。在给予PF-543 第7天后,未见典型自噬结构。HFE+CCl4组肝组织轻度水肿,膜完整、结构不清晰,粗面内质网数量丰富,明显扩张,表面附着核糖体颗粒较少。脂滴数量较少,局部融合、体积增大。该视野可见6个自噬溶酶体结构。在给予PF-543第7天后,该视野未见典型自噬结构(图6)。
3讨论
已有证据表明,SphK1/S1P信号在小鼠肺纤维化的病理生物学过程中具有重要作用[11],但其在肝纤维化中的具体分子机制尚不清楚,阻断SphK1的活化是否对肝纤维化具有保护作用。在本研究中,使用两种肝纤维化模型来评估SphK1抑制剂的保护作用。分别用CCl4和高脂乳剂联合CCl4造模,在暴露于两种不同的造模方式后,SphK1的抑制作用显著地减少了肝纤维化。但这种对肝纤维化的保护作用,随着肝纤维化病变程度的加重,不再显现。
SphK1靶向药物的设计和开发已成为国内外的研究热点。基于SphK1在炎性免疫相关疾病和肿瘤病理中的过度表达,大多数研究集中在SphK1选择性抑制剂的开发上。PF-543是目前应用最广泛的SphK1抑制剂,参与不同的细胞功能以改善疾病。既往一些研究[12-13]中,表明PF-543可以减少肺上皮细胞线粒体DNA损伤、募集单核细胞、降低活性氧的产生,以改善肺纤维化和肺损伤。对乳腺癌和炎症性肠病,包括胃肠道肿瘤的研究[14-15]表明,PF-543减少肿瘤微环境中各种免疫抑制因子的表达,以克服对免疫检查点抑制剂的耐药性。
本研究中,造模12周结束后肝脏病理结果显示造模成功,脂肪肝肝纤维化模型的纤维化程度明显重于CCl4组,达到肝硬化病理标准。第13周二次分组后各疾病阶段分别给予生理盐水或PF-543,共干预14天。干预第7天时,CCl4组、HFE+CCl4组纤维化面积均较模型组有改善。在干预第14天时,2组纤维化面积较第7天升高,与模型组相比无差异。Masson染色显示,PF-543对CCl4组病理改善程度更佳。HFE+ CCl4组由于纤维化程度更重,在干预前后病理未见明显差异(图4),提示PF-543在肝纤维化早期有一定治疗作用,但随着疾病程度加重,PF-543治疗效力逐渐减弱。运用Real-time PCR同时检测第1、7天SphK1及TGF-β、α-SMA等纤维化相关标志物的表达水平,模型组中SphK1随着纤维化程度加重,表达水平逐步降低;TGF-β、α-SMA等表达水平随着纤维化程度加重,表达水平逐步升高,干预后,上述指标未发生显著变化。表明在肝纤维化、肝硬化进程中,由SphK1主导的SphK/S1P信号处于抑制状态。有研究[16]表明,SphK1的活力和肝血窦的微循环状态密切相关,其活力增强时,肝窦微循环明显改善。其主要产物SIP可不同程度保护血管内皮细胞、改善微循环、减少白细胞的黏附以及抑制炎症诱导的血管通透性增加等病理学改变[17]。2020年开展的一项研究[4]表明,脂肪细胞特异性缺失SphK1导致糖耐量增加、脂肪细胞肥大和脂解受损,这意味着SphK1在脂肪细胞功能和维持器官整体代谢稳态中具有特定的作用。此外,在不同类型的细胞中,SphK1可能发挥截然相反的作用。在小鼠成纤维细胞、上皮细胞中特异性敲除SphK1基因,小鼠的肺纤维化程度明显缓解,胶原沉积量明显降低,纤维化相关指标TGF-β、Fn等蛋白水平明显降低。表明成纤维细胞中特异性敲除SphK1基因能够缓解肺纤维化进程。在上皮细胞中特异性敲除SphK1基因,发现上皮细胞中SphK1基因缺失后,小鼠的肺纤维化程度减轻,胶原沉积也明显减少。肺泡灌洗液中蛋白含量以及细胞数量也明显减少。纤维化相关指标Fn、α-SMA、Col1A2、TGF-β表达水平均下降。而在肺内皮细胞中SphK1则表现为具有抗炎和抗纤维化的作用[18]。这也表明SphK1/S1P信号在纤维化形成中可能具有双重作用[19-20],SphK1在上述细胞类型中的不同作用机制尚不清楚。
鉴于既往文献[21-22]对自噬与脂肪性肝病及纤维化关系相关性的探讨,本研究还检测了3种疾病模型中自噬标志物及自噬小体,发现SphK1与Atg5、Atg12、Becn1、Map1lc3a mRNA的表达均呈正相关,且Atg5、Atg12、Becn1的表达与肝纤维化面积呈负相关。提示SphK1可能调控细胞自噬,并影响了肝纤维化的状态。具体作用机制尚需功能验证实验。Atg蛋白是自噬的主要参与者,构成最重要的分子平台。Atg12-Atg5是自噬体膜上的标志性蛋白,这些蛋白正确连接可形成一种称作自噬体的细胞器。自噬体可像垃圾袋一样移除有毒物质。已知Atg5对脂肪细胞的自噬和脂肪生成具有重要功能[23]。本研究中,可以发现随着肝纤维化程度的加重,Atg5表达水平逐渐降低。Atg12与之变化趋势一致。同样,Becn1的水平与肝纤维化面积呈负相关。研究[24]表明,Becn1基因敲除小鼠血清脂联素水平降低,反之,Becn1自噬活跃可增强循环中具有生物活性脂联素水平,增强胰岛素敏感性,发挥非降解作用调节代谢稳态。Alexaki等[25]在2014年时即发现自噬与鞘脂代谢相关,鞘脂从头合成活跃可诱导自噬。同时,自噬控制着活体中鞘脂的水平,并表明在许多病理条件下,功能失调的自噬可能会导致鞘脂稳态的改变。本研究发现在肝纤维化与脂肪肝合并肝纤维化两种模型中,自噬活动均明显加强,而PF-543则对自噬有显著抑制作用。并同时伴有纤维化面积的减少。提示靶向SphK1可影响到肝脏的自噬水平,并进而缓解肝纤维化或代谢相关脂肪性肝病合并肝纤维化的状态。同时,本研究提示,无论对于肝纤维化还是脂肪肝合并肝纤维化,在疾病早期阶段,SphK1抑制剂均具有一定治疗作用,而如果肝纤维化程度过重,则治疗效应减弱,而用药疗程的问题也是今后值得探讨的一个方向。
倫理学声明:本研究方案于2020年12月11日经由新疆医科大学实验动物伦理委员会审批,批号:IACUC-20201211-13,符合实验室动物管理与使用准则。利益冲突声明:本文不存在任何利益冲突。作者贡献声明:王燕负责课题设计,资料分析,撰写论文;马丽负责动物实验;肖雄、雷冬梅、赵奉茹、郭力源参与收集数据,整理资料;王晓忠参与实验指导。
参考文献:
[1]DEVARAJ E, PERUMAL E, SUBRAMANIYAN R, et al. Liver fibrosis: Extracellular vesicles mediated intercellular communication in perisinusoidal space[J]. Hepatology, 2022, 76(1): 275-285. DOI: 10.1002/hep.32239.
[2]XUE R, FAN JG. Brief introduction of an international expert consensus statement: A new definition of metabolic associated fatty liver disease[J]. J Clin Hepatol, 2020, 36(6): 1224-1227. DOI: 10.3969/j.issn.1001-5256.2020.06.007.薛芮, 范建高. 代谢相关脂肪性肝病新定义的国际专家共识简介[J]. 临床肝胆病杂志, 2020, 36(6): 1224-1227. DOI: 10.3969/j.issn.1001-5256.2020.06.007.
[3]ESCUDERO-CASAO M, CARDONA A, BELTRAN-DEBON R, et al.Fluorinated triazole-containing sphingosine analogues. Syntheses and in vitro evaluation as SPHK inhibitors[J]. Org Biomol Chem, 2018, 16(39): 7230-7235. DOI: 10.1039/c8ob01867g.
[4]ANDERSON AK, LAMBERT JM, MONTEFUSCO DJ, et al. Depletion of adipocyte sphingosine kinase 1 leads to cell hypertrophy, impaired lipolysis, and nonalcoholic fatty liver disease[J]. J Lipid Res, 2020, 61(10): 1328-1340. DOI: 10.1194/jlr.RA120000875.
[5]AVNI D, HARIKUMAR KB, SANYAL AJ, et al. Deletion or inhibition of SphK1 mitigates fulminant hepatic failure by suppressing TNFα-dependent inflammation and apoptosis[J]. FASEB J, 2021, 35(3): e21415. DOI: 10.1096/fj.202002540R.
[6]LI L, WANG H, ZHANG J, et al. SPHK1 deficiency protects mice from acetaminophen-induced ER stress and mitochondrial permeability transition[J]. Cell Death Differ, 2020, 27(6): 1924-1937. DOI: 10.1038/s41418-019-0471-x.
[7]GENG T, SUTTER A, HARLAND MD, et al. SphK1 mediates hepatic inflammation in a mouse model of NASH induced by high saturated fat feeding and initiates proinflammatory signaling in hepatocytes[J]. J Lipid Res, 2015, 56(12): 2359-2371. DOI: 10.1194/jlr.M063511.
[8]LI Q, QIAN J, LI Y, et al. Generation of sphingosine-1-phosphate by sphingosine kinase 1 protects nonalcoholic fatty liver from ischemia/reperfusion injury through alleviating reactive oxygen species production in hepatocytes[J]. Free Radic Biol Med, 2020, 159: 136-149. DOI: 10.1016/j.freeradbiomed.2020.07.004.
[9]CHEN Y, AZAD MB, GIBSON SB. Methods for detecting autophagy and determining autophagy-induced cell death[J]. Can J Physiol Pharmacol, 2010, 88(3): 285-295. DOI: 10.1139/Y10-010.
[10]LAVIEU G, SCARLATTI F, SALA G, et al. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation[J]. J Biol Chem, 2006, 281(13): 8518-8527. DOI: 10.1074/jbc.M506182200.
[11]HUANG LS, NATARAJAN V. Sphingolipids in pulmonary fibrosis[J]. Adv Biol Regul, 2015, 57: 55-63. DOI: 10.1016/j.jbior.2014.09.008.
[12]CHERESH P, KIM SJ, HUANG LS, et al. The sphingosine kinase 1 inhibitor, PF543, mitigates pulmonary fibrosis by reducing lung epithelial cell mtDNA damage and recruitment of fibrogenic monocytes[J]. Int J Mol Sci, 2020, 21(16): 5595. DOI: 10.3390/ijms21165595.
[13]HA AW, SUDHADEVI T, EBENEZER DL, et al. Neonatal therapy with PF543, a sphingosine kinase 1 inhibitor, ameliorates hyperoxia-induced airway remodeling in a murine model of bronchopulmonary dysplasia[J]. Am J Physiol Lung Cell Mol Physiol, 2020, 319(3): L497-L512. DOI: 10.1152/ajplung.00169.2020.
[14]IMBERT C, MONTFORT A, FRAISSE M, et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1[J]. Nat Commun, 2020, 11(1): 437. DOI: 10.1038/s41467-019-14218-7.
[15]SUKOCHEVA OA, FURUYA H, NG ML, et al. Sphingosine kinase and sphingosine-1-phosphate receptor signaling pathway in inflammatory gastrointestinal disease and cancers: A novel therapeutic target[J]. Pharmacol Ther, 2020, 207: 107464. DOI: 10.1016/j.pharmthera.2019.107464.
[16]JIN LM, HUANG DS, LIU YX, et al. The effects of sphingosine kinase/1-phosphate sphingosine signaling on hepatic ischemia-reperfusion injury in rats[J]. Chin Exp Surg, 2020, 37(10): 1826-1829. DOI: 10.3760/cma.j.cn421213-20191031-00772.金麗明, 黄东胜, 刘原兴, 等. 鞘氨醇激酶及1-磷酸鞘氨醇信号参与大鼠肝窦微循环保护[J].中华实验外科杂志, 2020, 37(10): 1826-1829. DOI: 10.3760/cma.j.cn421213-20191031-00772.
[17]JIN LM, LIU YX, CHENG J, et al. The effect of SphK1/S1P signaling pathway on hepatic sinus microcirculation in rats with hepatic ischemia-reperfusion injury[J]. Hepatobiliary Pancreat Dis Int, 2022, 21(1): 94-98. DOI: 10.1016/j.hbpd.2021.06.003.
[18]HUANG LS, SUDHADEVI T, FU P, et al. Sphingosine kinase 1/S1P signaling contributes to pulmonary fibrosis by activating Hippo/YAP pathway and mitochondrial reactive oxygen species in lung fibroblasts[J]. Int J Mol Sci, 2020, 21(6): 2064. DOI: 10.3390/ijms21062064.
[19]KONO Y, NISHIUMA T, NISHIMURA Y, et al. Sphingosine kinase 1 regulates differentiation of human and mouse lung fibroblasts mediated by TGF-beta1[J]. Am J Respir Cell Mol Biol, 2007, 37(4): 395-404. DOI: 10.1165/rcmb.2007-0065OC.
[20]CENCETTI F, BERNACCHIONI C, NINCHERI P, et al. Transforming growth factor-beta1 induces transdifferentiation of myoblasts into myofibroblasts via up-regulation of sphingosine kinase-1/S1P3 axis[J]. Mol Biol Cell, 2010, 21(6): 1111-1124. DOI: 10.1091/mbc.e09-09-0812.
[21]LIAO YS, BAI LL. Role of exercise-induced autophagy in prevention and treatment of nonalcoholic fatty liver disease[J]. J Clin Hepatol, 2022, 38(5): 1156-1160. DOI: 10.3969/j.issn.1001-5256.2022.05.038.廖粤生, 白莉莉. 运动诱导细胞自噬在非酒精性脂肪性肝病防治中的作用[J]. 临床肝胆病杂志, 2022, 38(5): 1156-1160. DOI: 10.3969/j.issn.1001-5256.2022.05.038.
[22]LIU GJ, PENG L. Study on the role of autophagy in hepatic fibrosis[J]. Shandong Med J, 2018, 58(48): 108-110. DOI: 10.3969/j.issn.1002-266X.2018.48.031.刘国菊, 彭雷. 自噬在肝纤维化中作用的研究进展[J]. 山东医药, 2018, 58(48): 108-110. DOI: 10.3969/j.issn.1002-266X.2018.48.031.
[23]WANG X, WU R, LIU Y, et al. m6A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7[J]. Autophagy, 2020, 16(7): 1221-1235. DOI: 10.1080/15548627.2019.1659617.
[24]KURAMOTO K, KIM YJ, HONG JH, et al. The autophagy protein Becn1 improves insulin sensitivity by promoting adiponectin secretion via exocyst binding[J]. Cell Rep, 2021, 35(8): 109184. DOI: 10.1016/j.celrep.2021.109184.
[25]ALEXAKI A, GUPTA SD, MAJUMDER S, et al. Autophagy regulates sphingolipid levels in the liver[J]. J Lipid Res, 2014, 55(12): 2521-2531. DOI: 10.1194/jlr.M051862.
收稿日期:2022-11-25;錄用日期:2023-01-19
本文编辑:王莹
引证本文:WANG Y, MA L, XIAO X, et al. Mechanism of action of sphingosine kinase 1 inhibitor in a rat model of liver fibrosis[J]. J Clin Hepatol, 2023, 39(8): 1886-1894.
猜你喜欢
小学阅读指南·低年级版(2016年12期)2017-01-05 14:43:16
糖尿病新世界(2016年16期)2016-12-09 04:07:18
中国实用医药(2016年28期)2016-12-07 09:16:02
科技知识动漫(2016年7期)2016-07-29 20:50:17
上海医药(2016年13期)2016-07-26 22:56:06
上海医药(2016年13期)2016-07-26 22:54:16
上海医药(2016年13期)2016-07-26 22:52:38
科技视界(2016年11期)2016-05-23 08:10:09
小朋友·快乐手工(2015年12期)2016-01-14 00:29:00
幼儿园(2015年2期)2015-07-13 08:25:45
