
基于Yes相关蛋白/同源异型盒转录因子通路探讨淋巴管新生在高血压心肌重构中的作用
2023-04-13 01:24刘晶晶陈昌贵孙茹雪李立为
实用医学杂志 2023年3期
刘晶晶 陈昌贵 孙茹雪 李立为
湖北省武汉市中西医结合医院(武汉市第一医院)心血管内科重症监护室(武汉430022)
心脏肥大的发展最初被认为是应对容量或压力过载的一种适应性反应,但最终可能导致心力衰竭(heart failure,HF)[1]。尽管存在可以改善HF患者心脏功能障碍的治疗方法,但病死率仍然很高,迫切需要发现预防HF 的新目标或替代治疗策略。心脏有一个复杂的淋巴管网络,对维持组织液平衡、免疫细胞贩运和心脏功能至关重要。近年来,有研究注意到淋巴管功能损害参与压力过载引起的心脏功能障碍,而刺激心脏淋巴管生成可改善心脏淋巴运输和水肿,减少心脏炎症和纤维化,并改善左心室功能,但其中机制仍有待进一步研究[2-3]。哺乳动物Hippo 信号通路是心血管系统中细胞命运、细胞分化、增殖、迁移和器官形态发生的重要调节者[4]。Hippo 通路的核心涉及大肿瘤抑制激酶1 和2、Yes相关蛋白(Yes-associated protein,YAP)及其旁系同源物TAZ。最近发现,YAP 在血管生长和淋巴管生长的内皮细胞中高度富集,并在萌芽血管生成和血脑屏障成熟中发挥促进作用[5]。重要的是,研究发现YAP 通过调节同源异型盒转录因子(prospero-related homeobox domain 1,PROX1)表达参与淋巴内皮细胞可塑性[6]。然而,YAP/PROX1 通路是否调节压力过载引起的心脏淋巴管功能损害修复仍不清楚。因此,本研究利用YAP 基因敲除小鼠检查了YAP/PROX1 通路在横主动脉收缩(transverse aortic constriction,TAC)诱发的心脏肥大和功能障碍中的潜在作用。同时,检查了AngⅡ刺激的原发性小鼠淋巴管内皮细胞(lymphatic endothelial cells,LECs)的内皮细胞功能和淋巴管生成能力。
1 材料与方法
1.1 动物和分组处理20 只野生型(WT)小鼠和20 只YAP 基因敲除(YAP-KO)小鼠(8 周龄、雄性)均购自均购自杰克森艾特生物科技(北京)有限公司。实验动物许可证号:SCXK(京)2019-0004。将YAP-KO 小鼠随机分为两组:假手术(Sham)+YAPKO 组(n= 10)和TAC+YAP-KO 组(n= 10),和将WT 小鼠随机分为两组:Sham+WT 组(n= 10)和TAC+WT 组(n= 10)。除Sham+YAP-KO 组和Sham+WT 组外,其他组建立6 周TAC 模型。
参照文献方法,通过TAC 手术诱导小鼠心脏肥大和HF 模型的持续压力过载[7]。小鼠用氯胺酮(0.2 g/kg)和赛拉嗪(0.01 g/kg)进行腹腔注射麻醉。在充分暴露横主动脉后,将6-0 尼龙缝合线置于锁骨和左颈动脉之间,将27 号钝针置于横主动脉处,并在及时拔出针头后迅速打两个结以产生65%~70%的收缩力。最后,用4-0 PROLENE 缝线缝合皮肤。对于Sham 小鼠,除结扎外,所有操作都以相同的程序进行。
1.2 超声心动图和血压测量在TAC手术后的6周,使用30 MHz 探头进行超声心动图检查。测量左心室射血分数(LVEF)、分数缩短(FS)、舒张末期左心室前壁尺寸[LVAW(d)]、舒张末期左心室后壁尺寸[LVPW(d)]、舒张末期的左心室内部尺寸[LVID(d)]、收缩末期的左心室前壁尺寸[LVAW(s)]、收缩末期的左心室后壁尺寸[LVPW(s)]、收缩末期的左心室内部尺寸[LVID(s)]。通过创尾套系统(美国Softron 公司)测量小鼠的收缩压(SBP)、平均血压(MBP)、舒张压(DBP)和心率(HR)。
1.3 心脏水含量测量取出小鼠心脏的心房和大血管以评估心室的湿重。心室在65 ℃下干燥5 d后,获得心脏干重。心肌含水量按以下公式计算:心脏含水量=(心脏湿重-心脏干重)/心脏湿重×100%。
1.4 组织病理学和免疫染色心脏组织在室温下用4%多聚甲醛在磷酸盐缓冲盐水中固定48 h并嵌入石蜡中。将心脏组织切成5 μm 的切片用于以下分析。切片用小麦胚芽凝集素(WGA)染色以测量心肌细胞的大小。用Image J 对心肌细胞面积进行了量化。将载玻片与特异性淋巴管内皮细胞透明质酸受体1(lymphatic vessel endothelial receptor-1,LYVE1)(稀释度,1∶200)的一级抗体在4 ℃下孵育过夜,进行免疫组织化学。次日,与二抗在37 ℃下孵育30 min,检测LYVE1 的表达。
1.5 细胞培养小鼠LECs 购自美国Procell 公司,在含有5% 胎牛血清和1% 青霉素-链霉素的DMEM 基础培养基中生长,温度为37 ℃。在培养基中使用适当剂量的AngⅡ(10-6mol/L)处理48 h,对LECs 的功能和基因表达情况进行检测。为了抑制YAP 在体外的表达,用50 nmol/L 的YAP小干扰RNA(si-YAP)转染LECs 48 h。si-YAP 的序列是5′-GGAUGUAGCUGAGCUGCUUTTAAGCAGCUCA-GCUACAUCCTT-3′。为了在体外过量表达YAP、PROX1,使用YAP、PROX1 质粒转染细胞。在转染48 h 后收获转染了质粒或siRNA 的细胞并进行功能分析。LECs 分为以下6 组进行:对照(Control)组、AngⅡ+NC 组、AngⅡ+si-YAP 组、AngⅡ+YAP 组、AngⅡ+si-YAP+Vector 组和AngⅡ+si-YAP+PROX1 组。其中,Control 组细胞加入空白溶剂进行处理,AngⅡ+NC 组细胞加入si-NC、Vector转染,其他组加入对应的小干扰RNA 或质粒进行处理。si-NC、si-YAP,YAP、PROX1 质粒和相应载体(Vector)均购自美国Ambion 公司。
1.6 伤口愈合试验将处理后的LECs 培养在6 孔板中并形成单层。用黄色吸头在单层LECs 表面进行1 mm 宽的线性划痕,然后用含或不含AngⅡ的无血清培养基替换培养基,持续24 h。在随机选择的5 个区域测量相对愈合距离,并使用Image J 进行分析。
1.7 EDU 染色LECs 以每孔103~104个细胞的密度被接种到96 孔板中。将细胞在含有EDU 的培养基中培养24 h,并用4%的聚甲醛固定。用Apollo 标记EDU 阳性细胞,用Hoechst33342 标记细胞核。在荧光显微镜(日本Olympus 公司)下观察EDU 阳性细胞。
1.8 Proteome ProfilerTM 阵列应用小鼠血管生成阵列试剂盒(美国R&D Systems 公司)进行因子筛选。收集细胞培养上清液。将阻断缓冲液加入到膜上,在摇板上摇晃1 h。将样品与15 μL 重组的检测抗体鸡尾酒混合,然后在室温下孵育1 h,将混合物转移到膜上孵育过夜。用链霉蛋白-HRP缓冲液孵育细胞30 min。通过加入Chemi Reagent混合液进行最终检测,然后进行扫描。
1.9 PCR 分析用RNAeasy Plus 微型试剂盒(美国QIAGEN 公司)分离细胞的总RNA。使用RNA转cDNA 预混合液(Clontech Laboratories)将分离的RNA 转录成cDNA。使用SYBR Green 试剂(瑞士Roche Diagnostics 公司)在StepOnePlus 系统(美国Life Technologies 公司)上进行实时PCR 反应。在所有实验中,GAPDH 被用作参考基因,以使基因表达正常化。引物序列如下:ANG,正向5′-TGCCACAACTCATCCGACA-3′,反向5′-TCTGTTGCGGGTTTGAGT-3′;VEGF,正向5′-CCTGCCATACGCCACATCAT-3′,反向5′-TGCATGAAGCCATCCTTC-3′;MMP-9,正向5′-ATGGTTCTGCTCCTGGTAA-3′,反向5′-AGGTCTGCCTGCATTTCT-3′;IGFBP-3,正向5′-GAATGGCAGTGTGGACCTCT-3′和反向5′-CAGCCTCAAACTCCACCATT-3′;PROX1,正向5′-AAAGCAAAGCTCATGTTTTTATACC-3′和反向5′-GTAAAACTCACGGAAATTGCTAAACC-3′;GAPDH,正向5′-AGGTCGGTGTGAACGGATTTG-3′,反向5′-TGTAGACCATGTAGTTGAGGTCA-3′。
1.10 蛋白质印迹分析使用含有蛋白酶混合抑制剂和磷酸酶混合抑制剂的放射免疫沉淀试验缓冲液(瑞士Roche Diagnostics公司)制备细胞裂解液。取20 μg 的总蛋白通过4%~12% Bis-Tris 凝胶(美国Life Technologies 公司)解析,并转移到聚偏二氟乙烯膜(美国Thermo Fisher Scientific 公司)。在4 ℃下将膜与以下针对指定靶蛋白的一抗进行探测:抗VEGFR3 抗体(1∶1 000)、抗LYVE1 抗体(1∶1 000)和抗GAPDH 抗体(1∶5 000)。随后,将膜与过氧化物酶偶联的抗小鼠或抗兔IgG 抗体一起孵育90 min。使用Western ECL 底物(美国Bio-Rad Laboratories)进行免疫印迹,并通过Image J 测量条带的强度。
1.11 统计学方法所有结果均以均数±标准差表示。使用SPSS 22.0 进行统计分析。所有实验数据均呈正态分布,并使用单向方差分析多组间的差异,然后用Fisher 最小显著性差异检验进行事后配对比较。P<0.05 认为差异有统计学意义。
2 结果
2.1 YAP 缺失加重了TAC 诱导的高血压心脏功能障碍与Sham+WT 组相比,TAC+WT 组小鼠EF%、FS%、LVAW 厚度和LVPW 厚度显著下降(P<0.05),和SBP、DBP、MBP、LVID 厚度显著增加(P<0.05)。TAC+YAP-KO 组小鼠EF%、FS%、LVAW 厚度和LVPW 厚度显著低于TAC+WT 组(P<0.05),并且LVID 厚度显著增加(P<0.05,图1、表1)。
表1 WT 和YAP-KO 小鼠在TAC 手术6 周后或假性对照后的血压和超声心动图参数Tab.1 Blood pressure and echocardiographic parameters of wild-type WT and YAP-KO mice after TAC operation for 6 weeks or sham control ±s
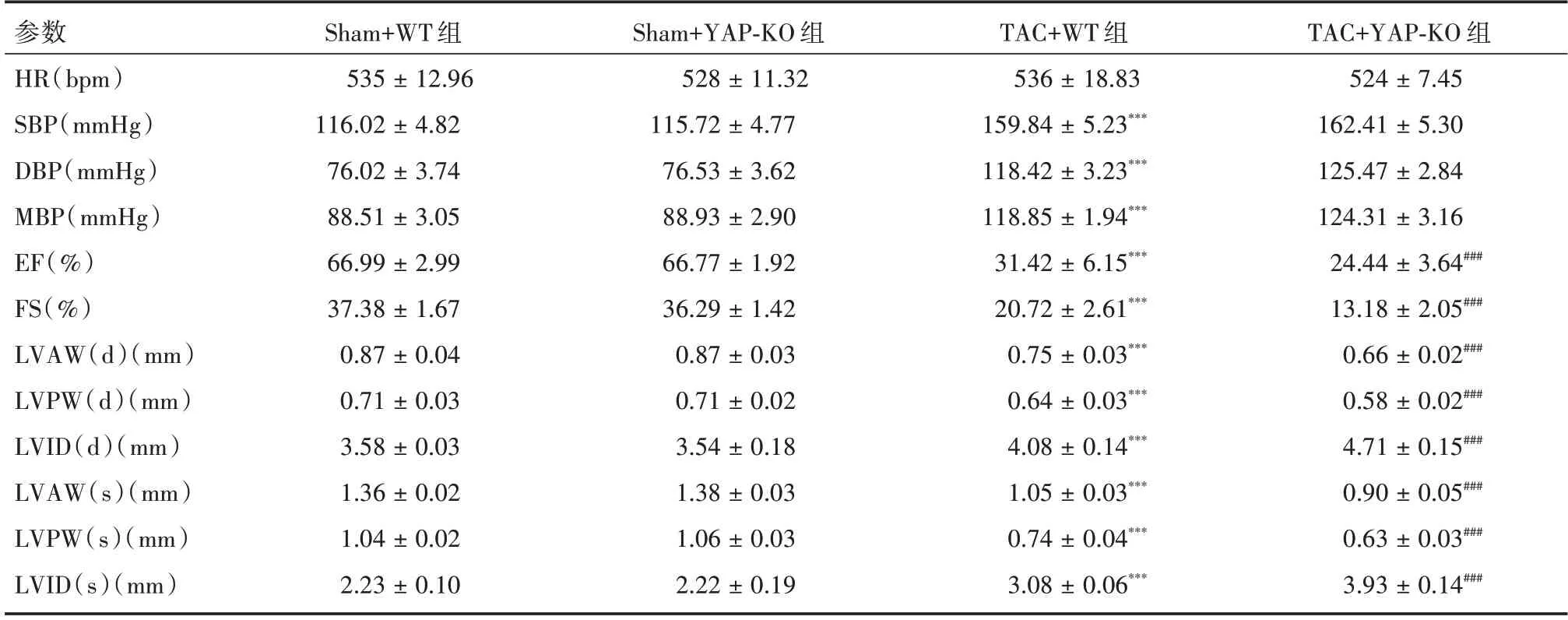
表1 WT 和YAP-KO 小鼠在TAC 手术6 周后或假性对照后的血压和超声心动图参数Tab.1 Blood pressure and echocardiographic parameters of wild-type WT and YAP-KO mice after TAC operation for 6 weeks or sham control ±s
注:与Sham+WT 组比较,***P <0.001;与TAC+WT 组比较,###P <0.001
参数HR(bpm)SBP(mmHg)DBP(mmHg)MBP(mmHg)EF(%)FS(%)LVAW(d)(mm)LVPW(d)(mm)LVID(d)(mm)LVAW(s)(mm)LVPW(s)(mm)LVID(s)(mm)Sham+WT 组535 ± 12.96 116.02 ± 4.82 76.02 ± 3.74 88.51 ± 3.05 66.99 ± 2.99 37.38 ± 1.67 0.87 ± 0.04 0.71 ± 0.03 3.58 ± 0.03 1.36 ± 0.02 1.04 ± 0.02 2.23 ± 0.10 Sham+YAP-KO 组528 ± 11.32 115.72 ± 4.77 76.53 ± 3.62 88.93 ± 2.90 66.77 ± 1.92 36.29 ± 1.42 0.87 ± 0.03 0.71 ± 0.02 3.54 ± 0.18 1.38 ± 0.03 1.06 ± 0.03 2.22 ± 0.19 TAC+WT 组536 ± 18.83 159.84 ± 5.23***118.42 ± 3.23***118.85 ± 1.94***31.42 ± 6.15***20.72 ± 2.61***0.75 ± 0.03***0.64 ± 0.03***4.08 ± 0.14***1.05 ± 0.03***0.74 ± 0.04***3.08 ± 0.06***TAC+YAP-KO 组524 ± 7.45 162.41 ± 5.30 125.47 ± 2.84 124.31 ± 3.16 24.44 ± 3.64###13.18 ± 2.05###0.66 ± 0.02###0.58 ± 0.02###4.71 ± 0.15###0.90 ± 0.05###0.63 ± 0.03###3.93 ± 0.14###
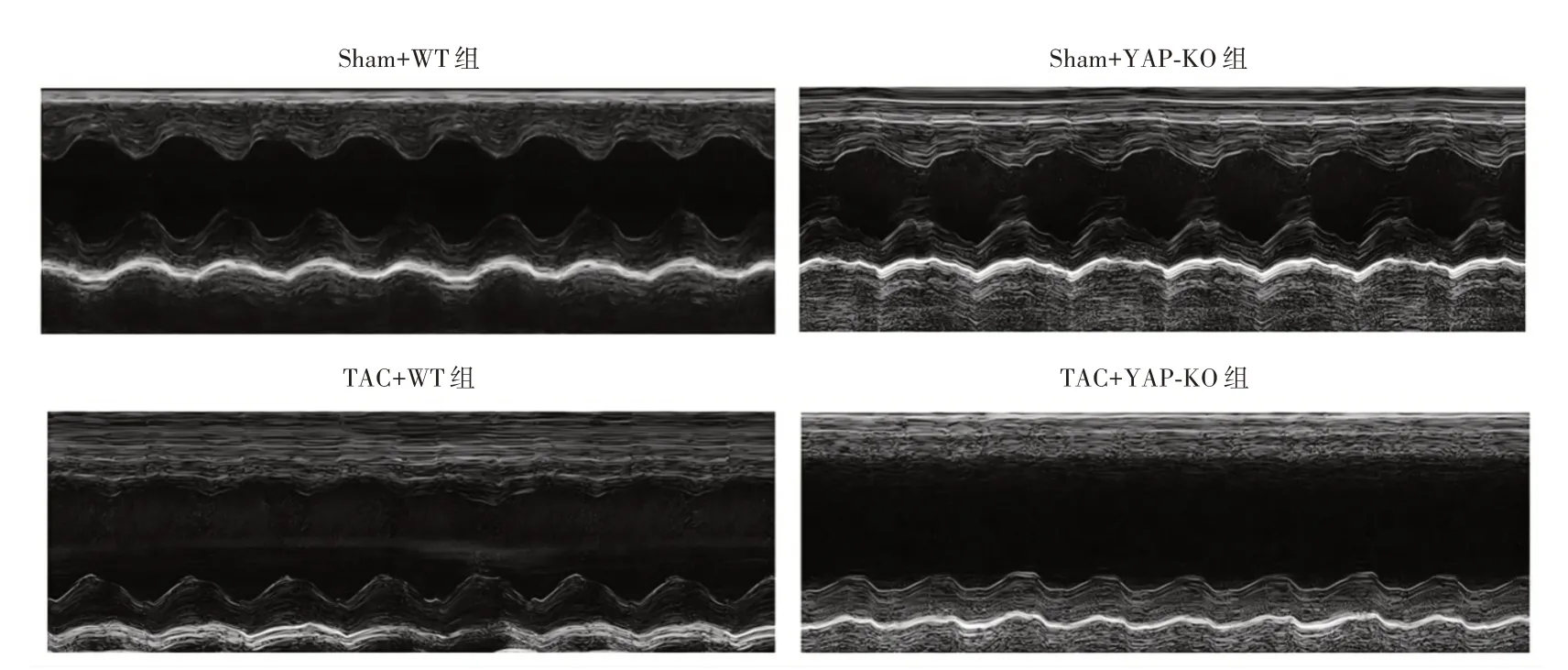
图1 各组代表性的M 型组织多普勒超声心动图Fig.1 Representative M-mode tissue Doppler echocardiography of each group
2.2 YAP 缺失加重TAC 诱导的心脏肥大和纤维化与Sham+WT 组相比,TAC+WT 组小鼠心脏质量比、心肌细胞大小和间质纤维化面积均显著增加(P<0.05)。与TAC+WT 组相比,TAC+YAP-KO组小鼠心脏质量比、心肌细胞大小和间质纤维化面积均显著增加(P<0.05)。
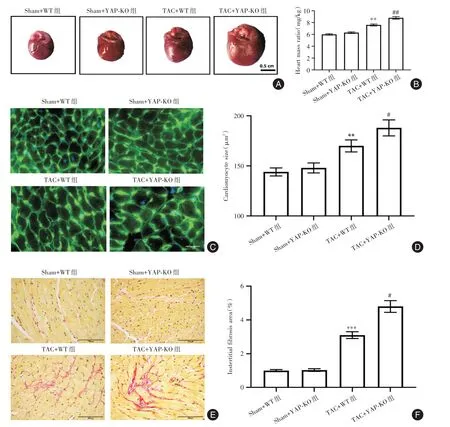
图2 YAP 缺失加重TAC 诱导的心脏肥大和纤维化Fig.2 YAP deletion aggravates cardiac hypertrophy and fibrosis induced by TAC
2.3 YAP 是维持心脏淋巴管生成的必要条件与Sham+WT 组相比,TAC+WT 组小鼠淋巴微血管密度显著降低(P<0.05),并且TAC+YAP-KO 组小鼠淋巴微血管密度较TAC+WT 组进一步降低(P<0.05)(图3A)。此外,与Sham+WT 组相比,TAC+WT 组小鼠心脏水肿及心脏组织中YAP 蛋白表达显著增加(P<0.05),和血清VEGFC 水平以及心脏组织中VEGFR3、LYVE1 蛋白表达显著降低(P<0.05),并且TAC+YAP-KO 组小鼠上述指标的变化程度较TAC+WT 组更大(P<0.05)(图3B-F)。

图3 YAP 的缺乏降低了TAC 诱导后的心脏淋巴管生成Fig.3 Lack of YAP reduces cardiac lymphangiogenesis induced by TAC
2.4 YAP 调节LECs 的功能如图4A 所示,与对照组相比,AngⅡ+NC 组愈合面积显著下降(P<0.05),并且AngⅡ+si-YAP 组下降程度较AngⅡ+NC 组更大(P<0.05),而AngⅡ+YAP 组的愈合面积较AngⅡ+NC 组显著增加(P<0.05)(图4A-B)。进行EDU 染色试验来检查增殖。如图4C、D 所示,与对照组相比,AngⅡ+NC 组LECs 的增殖显著下降(P<0.05),并且AngⅡ+si-YAP 组下降程度较AngⅡ+NC组更大(P<0.05),而AngⅡ+YAP组LECs的增殖较AngⅡ+NC 组显著增加(P<0.05)。LECs的管形成检查显示,与对照组相比,AngⅡ+NC 组LECs 的管形成数量显著下降(P<0.05),并且AngⅡ+si-YAP 组下降程度较AngⅡ+NC 组更大(P<0.05),而AngⅡ+YAP 组LECs 的管形成数量较AngⅡ+NC 组显著增加(P<0.05,图4E-F)。
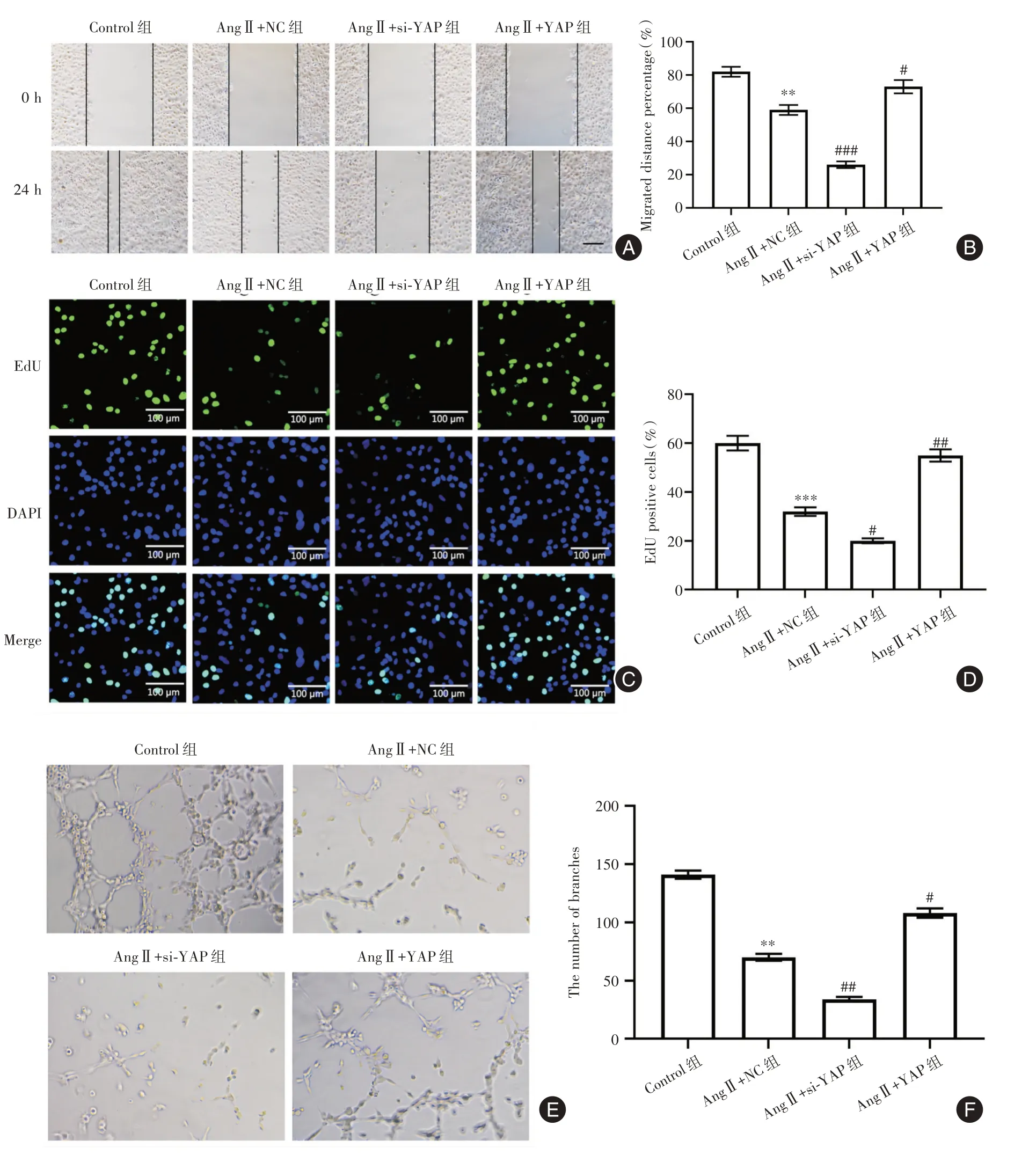
图4 YAP 调节LECs 的迁移、增殖和管形成Fig.4 YAP regulates the migration,proliferation and tube formation of LECs
2.5 YAP 上调LECs 中PROX1 表达以加速淋巴管生成为了证实YAP 对LECs 影响的分子机制,研究应用小鼠血管生成阵列试剂盒对潜在的生长因子进行了筛选。发现了5 个顶级的分化因子:ANG、VEGF、MMP-9、PROX1 和IGFBP-3(图5A)。然后利用PCR 进行验证,结果显示只有PROX1 水平在YAP 敲低后显著下调(图5B)。此外,与AngⅡ+si-YAP+Vector 组相比,AngⅡ+si-YAP+PROX1组LECs 的愈合面积、增殖细胞和管形成数量显著增加(P<0.05,图5C-H)。

图5 YAP 上调LECs 中PROX1 表达以加速淋巴管生成Fig.5 YAP upregulates PROX1 expression in LECs to accelerate lymphangiogenesis
3 讨论
慢性压力过载引起了与心脏纤维化和功能障碍有关的病理性肥大[8]。已发现多种分子事件和信号通路参与了这一过程,但确切的机制还需要探索。微循环,尤其是淋巴微血管循环,在保护心脏不受伤害方面起着重要作用[8]。心脏淋巴功能障碍发生在各种心血管疾病中,影响心肌液体平衡和炎症,可能诱发血管功能障碍、心脏重塑和缺血应激后心脏功能障碍[9]。这项研究证明了YAP/PROX1 信号通路在心肌淋巴管生成中的重要性,这是从压力过载诱导的肥厚过渡到HF 的一个新的调节途径。YAP 敲除加重了压力过载诱导的心脏淋巴管生成的减少,导致心脏水肿、肥大重塑和功能障碍。因此,本研究数据支持YAP 在持续压力过载后心脏重塑和功能障碍的发病机制中发挥作用。
YAP 是Hippo 信号传导途径的主要下游效应器[10-11]。研究报道,YAP 信号通路参与了胚胎发育、组织再生和体细胞干细胞的平衡[12]。最近,YAP 因其在病理性淋巴管发展中的作用而引起越来越多的关注。在各种癌症中,包括胃癌和非小细胞肺癌,YAP 异常激活与TNM 分期和淋巴转移明显相关[13]。关于淋巴管生成,YAP 通过调控PROX1 的表达,在重塑淋巴丛模式和产后淋巴瓣的维持中发挥促进作用[14]。此外,YAP 抑制剂verteporfin 不仅能减少淋巴管介导的肿瘤转移,还通过促进淋巴消退应用于角膜移植后的免疫排斥[15-16]。本研究中,TAC 应激后YAP-KO 小鼠比WT 小鼠表现出更显著的HF 特征。此外,使用LYVE1 作为LECs 的标记,本研究数据进一步表明,YAP-KO 引起的异常可能是由于淋巴微血管的密度下降造成。YAP 通过调节氧化应激和代谢重编程保护心脏免受内皮功能障碍的影响[17-18]。在本研究中,AngⅡ降低了LECs 的迁移、增殖和管形成,抑制YAP 可加剧降低程度,但YAP 过表达后则逆转这些变化。本研究结果与ZHONG 等[19]报道的结果一致,在他们的研究中YAP 的激活促进了LECs 在感染后的增殖、侵袭和管形成。这些结果表明YAP 通过介导淋巴管的生成参与了TAC 诱导的心脏损伤的发展。
淋巴转移的一个重要过程是局部淋巴管生成。研究人员发现,心肌细胞和淋巴内皮细胞之间的串联是淋巴管生成的一个重要部分,生长因子可能是它们之间的信使[9,20]。本研究应用小鼠血管生成阵列试剂盒对潜在的生长因子进行了筛选。我们选择血管生成阵列试剂盒出于以下原因:(1)一些细胞表面受体家族,如整合素α4β1 和α2β1[21-22],已被报道为血管生成和淋巴管生成的调节因子,表明血管生成相关蛋白也可能对淋巴管生成有类似的影响。(2)本阵列试剂盒中包括的一些血管生成相关的生长因子,如ANG1、ANG2、PROX1、VEGF-C 和VEGF-A,被证明在淋巴管生成中起作用[23-24]。根据血管生成相关因子的表达,我们发现PROX1 与YAP 调节的淋巴管生成有关。PROX1 是主转录因子,控制着LECs 的分化和维持淋巴的完整性。与先前研究一致[6,25],本研究证实了YAP/PROX1 信号通路在淋巴管生成中的调节作用,并且PROX1 上调有效逆转了YAP 敲低对LECs 愈合、增殖和管形成的负面影响。因此,YAP/PROX1 信号通路可能是治疗压力过载引起的心脏功能障碍的潜在靶点。
总之,本研究确定了YAP/PROX1 信号通路是压力过载诱导的心脏淋巴管生成、适应性肥大和HF 的关键调节因素。需要进一步的调查来阐明YAP 参与调节LECs 中PROX1 表达的分子机制,并测试YAP/PROX1 信号传导在其他动物模型中预防心脏肥大和HF 的效果。
【Author contributions】Liu Jingjing performed the experiments and wrote the article.Chen Changgui and Sun Ruxue performed the experiments.Li Liwei designed the study and reviewed the article.All authors read and aplproved the final manuscript as submitted.
猜你喜欢
昆明医科大学学报(2021年4期)2021-07-23
世界最新医学信息文摘(2021年12期)2021-06-09
江苏卫生保健(2020年9期)2020-10-23
中国临床医学影像杂志(2019年4期)2019-06-18
国际呼吸杂志(2019年4期)2019-03-12
听力学及言语疾病杂志(2015年5期)2015-12-24
哈尔滨医药(2015年3期)2015-12-01
医学研究杂志(2015年4期)2015-06-10
中国当代医药(2015年16期)2015-03-01
组织工程与重建外科杂志(2011年2期)2011-03-27
