
腹膜后Castleman病1例报告并文献复习
2023-03-14 06:29江书典江敏芝胡明政
中国医药科学 2023年4期
江书典 江敏芝 胡明政
三峡大学第一临床医学院 湖北省宜昌市中心人民医院肝胆胰外科,湖北宜昌 443003
Castleman病(Castleman disease,CD)又称巨淋巴结增生或血管滤泡性淋巴样增生,是临床上一种较为稀少的、表现多样的淋巴组织异常增生性疾病。在2020年5月国家公布的第一批罕见病目录中,就有关于此病的报道[1]。该病于1956年由美国病理学博士Castleman等[2]第一次报道并予以命名。目前国内外报告的文献并不多见,大多以病例的形式展现,鉴于我们对此病的研究不够深入,因此临床上出现误诊及漏诊的情况时有发生,本文回顾性分析1例腹膜后CD的临床资料,供临床参考。
1 病例资料
患者,女性,56岁。因“上腹部疼痛5 d”于2021年5月10日就诊。患者无其他伴随症状,患病以来体力稍有下降,体重短期内无明显改变,入院后查血三系(白细胞2.4×109/L,红细胞3.6×1012/L,血小板93×109/L)均提示下降,肿瘤标志物:甲胎蛋白、癌胚抗原、糖类抗原稍有所上升。胰腺平扫增强(CTA+CTV)检查示:胃-胰尾之间占位伴钙化,考虑间叶源性①胃肠腔外间质瘤,②神经源性肿瘤或血管瘤。肝脏多发海绵状血管瘤。胃左动脉供血胃-胰间肿瘤。肝固有动脉供血肝右叶及部分肝左内叶,胃左动脉分支供血肝左外叶,胃冠状静脉曲张伴静脉瘤形成。综合上述检查,初步诊断为腹膜后肿瘤。完善相关术前检查及排除手术禁忌后于2021年5月13日在全身麻醉下行腹腔后肿瘤切除术,术中见肿瘤位于胃后壁下方,胰腺上缘,大小约为6 cm×5 cm,血供丰富,边界清楚,质地稍硬,活动尚可,包膜完整,未见周围淋巴结肿大。利用超声刀及电刀游离腹膜后肿瘤,分离与胃后壁及周围组织粘连,游离肿瘤至基底部后,结扎基底部血管,完整切除病灶。术后病检:(大体观)暗红色不整形组织一块,大小8.2 cm×7.5 cm×3.1 cm,局部具包膜,切面灰红,实性,质中。病理诊断:(腹膜后病损)镜下显示淋巴结结内淋巴滤泡大多散在分布,其结构混乱,其生发中心萎缩,套区增宽呈同心圆状,局部可见血管长入,滤泡间区可见丰富的小血管增生伴管壁玻璃样变性。免疫组化显示:CD3(滤泡间+),CD20(滤泡+),Ki-67(低表达),CD10(个别萎缩生发中心+),Bcl-2(+),CD5(滤泡间+),CyclinD1(-),SOX-11(-),CD38(散在+),CD138(散在+),HHV8(-),分子病理结果:EBER(-)。见图1~2。诊断为腹膜后淋巴结CD,透明管型。患者术后恢复可,于2021年5月21日出院,出院1年后予以电话随访,患者未诉特殊不适。
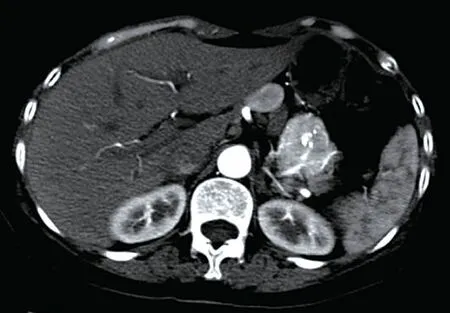
图1 CT增强
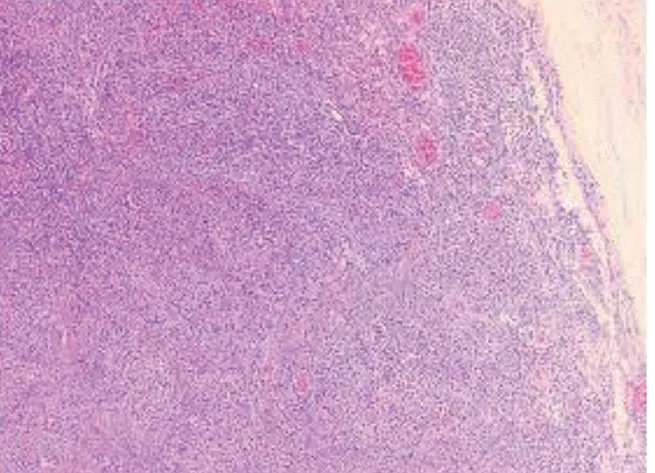
图2 HE染色(100×)
2 讨论
CD是临床上一种较为罕见的淋巴组织增生性疾病,相关文献显示,CD每年的患病率约为1/50 000[3],儿童、青年及老年人均可发病,且男性和女性发病率相当[4],该病可发生于全身的任何部位(包括淋巴结、结外组织和器官),有关研究数据表明其最好发于纵隔,颈部、腹部、腹股沟区等部位次之,腹膜后发病率相对较低[5],临床上根据淋巴结累及情况将CD分为单中心型(unicentric CD,UCD)和多中心型(multicentric CD,MCD)两种类型,其中单中心型约占80%,常发生于25岁左右的人群,累及单个淋巴结区域,无明显症状,多数由体检时发现,远期生存率高[6]。多中心型发生率相对较低,常发生于50岁左右的人群,可累及多个淋巴结区域及一些重要脏器(肝、肺、肾等),多有全身症状,病死率较高[7]。
CD的病因及其发病机制目前尚不明确,根据现有的研究结果显示[8],不论是UCD还是MCD,白细胞介素6(IL-6)表达变化及人疱疹病毒8(HHV-8)感染,两大主要发病机制目前研究较为明确,不管是以往还是现有的研究证据表明IL-6与CD联系紧密。以往研究证实CD患者的血清标本和淋巴结标本中IL-6表达增强,切除肿大淋巴结前后对比发现,IL-6的水平较前明显降低,近些年研究发现CD患者存在多种形态的IL-6受体。这些将会为未来精准治疗该病提供重要方向。HHV-8又称卡波西肉瘤疱疹病毒,HHV-8感染与CD相关的可能机制: ①病毒编码IL-6类似物,刺激B细胞增殖并抑制凋亡,从而增加IL-6的表达。②诱导血管内皮生长因子的表达,继而促进新生血管形成。两者共同推动病情进展。研究显示,除此之外还有一些等待深入研究的发病机制,如IL-6以外的其他细胞因子(VEGF、IL-1/IL-5/IL-10、肿瘤坏死因子、表皮生长因子受体等)以及HHV-8以外其他病毒感染(巨细胞病毒、EB病毒等)[9]。
CD的临床表现多无特异性,对于UCD患者,大多数均因体检发现发病部位孤立的肿大淋巴结,或因肿瘤较大压迫周边脏器产生相应症状而就诊,姚静等[10]报道1例儿童腹股沟区CD,因“发现左侧腹股沟区肿物2月”而就诊,刘修平等[11]报道1例左侧颈部CD,因“发现左侧颈部淋巴结肿大2月”而入院,在一项404例的回顾性临床研究当中发现,单个肿大的淋巴结直径约为5.5 cm[12]。本例患者因上腹部疼痛5 d就诊,疑似病变压迫周边组织,术中见肿瘤大小约为6 cm×5 cm,与文献报道基本相符。对于MCD的患者则表现出多样性,除了表现出全身多发的肿大淋巴结以外,还可表现出(如发热、贫血、盗汗、腹胀腹泻等)一般表现及重要器官损害(肝脾肿大、肝肾损伤等)等表现[13]。病情严重者还可表现出多种合并症[14],如:①POEMS综合征,常累神经、内分泌、皮肤等多个系统,与浆细胞有关;②副肿瘤性天疱疮,临床上较为罕见,常以损害黏膜为特征,特别是口腔黏膜,表现为黏膜糜烂及皮肤多发皮疹等,除此之外还有一些较为少见的并发症如TAFRO综合征、淋巴瘤等。
CD的影像学表现同样也无特异性,B超、CT、MRI、PET-CT等对疾病诊断起一定的辅助作用,CD在B超下表现为边缘清晰、形态规则、内部以低回声为主的实性肿块,何艳萍等[15]回顾性分析了34例CD患者的超声特征,总结出若在B超下出现肿物内部断续短线状高回声,应高度怀疑此病。但这仅限于体表肿大的淋巴结的判断,CT表现常无明显特征性,多数病灶表现出斑片状或分支样钙化,少数病灶表现出低密度或囊性改变,有些学者[16]认为钙化是CD的重要特征性改变,但这又限于UCD患者,相比于B超及CT,MR在软组织上更具有优势,在T2WI抑脂下表现为高低混合信号,T1WI抑脂下表现出较为均一的稍低信号,增强扫描动脉期强化明显,平扫低信号区几乎不强化,其在抑脂T2WI像及DWI序列病灶呈高信号的表现有一定的特征性[17]。PET-CT对于淋巴结的代谢判断方面较有优势。是该病与淋巴瘤鉴别的一个重要依据。CD最终确诊需依靠病检及免疫组化,其病理特点表现多样,常表现出3种类型,分别为透明血管型(HV) 、浆细胞型(PC)、混合型(Mix),其中(HV)约占总类型的90%,最为多见,浆细胞型(PC)次之,混合型(Mix)则占比最少。HV型镜下多可见典型的“洋葱皮”样或“棒棒糖”样结构,即增生的小淋巴细胞围绕着萎缩的生发中心呈同心圆样排列,萎缩的生发中心内充满异常增生的滤泡树突细胞,淋巴滤泡间有玻璃样或纤维样变性的小血管穿入生发中心,而PC型常不表现出类似“洋葱皮”样结构,但可见成簇的不同程度浆细胞增长,混合型则兼具两者的部分结构。就免疫组化而言,三者不表现出明显差异,徐傲等[18]学者回顾性分析45例CD患者的免疫组化发现,CD20、CD3、bcl-2、CD23、CD10、CD4、CD68、CD38、CD138、Ki-67 均有不同比例的表达,本例患者免疫组化结果基本与报道相符。
对于CD的诊断,最早在20世纪90年就有学者提出该病的诊断标准[19],见表1。此诊断目前仍在广泛使用,之后有学者提出该病的金标准[20]即术前淋巴结活检,术中快速病检,术后常规病检及免疫组化。因该病术前诊断较为困难,容易误诊,故常常需要跟一些疾病相鉴别,UCD患者病变绝大多数均表现为单一肿大淋巴结,本例患者为腹膜后CD,因此需与嗜铬细胞瘤、副神经节瘤、腹部血管性肿瘤等相鉴别,对于MCD,因其病变累及全身,特别注意应该与一些能引起全身多处淋巴结肿大的恶性肿瘤相鉴别。诸如多发性骨髓瘤,淋巴瘤等。
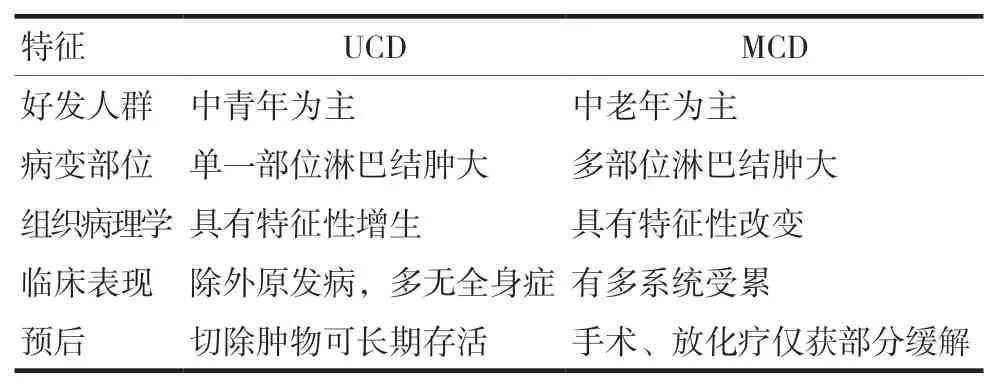
表1 CD诊断标准
根据以往的研究总结认为,对于绝大多数的UCD患者,手术完整切除病灶为该病主要的治疗手段,且能起到一定的诊断作用,对于少数术前评估无法切除病灶或术中难以完整切除病灶的患者,有学者曾以个案报道,予以行术前新辅助化疗,能使病灶缩小而后根治性手术切除[21]。而术中难以完整切除病灶的患者,术后给予适当的化疗也能起到一定的疗效[22]。有文献报道显示[23],UCD患者对放疗的敏感性高达72%,以往的手术方式均为开放性手术切除,随着微创时代的发展,腹腔镜技术也被应用于该病的治疗,近年来陆续出现一些行后腹腔镜手术成功的案例报道,腹腔镜手术创伤小,恢复快,目前认为是后腹膜CD理想的治疗方法。本例患者术前诊断不明确,故采用传统开放的手术方式,术后病检提示腹膜后CD。由于MCD患者表现多样,且累及全身,故目前为止仍无标准的治疗方案,仍在持续的探索中,对于早期病灶且只累及少数区域的患者,可选择手术治疗及术后放疗的方式[24]。目前MCD患者大多采用保守维持方式治疗为主,①对于那些没有全身反应而仅有全身多处淋巴结肿大的患者,即便是不经过系统治疗,少数患者病情也可以不发生变化,这样的患者可予以等待和观察;②激素治疗能够起到一定的缓解作用,Dispenzieri等[7]研究报道显示一半的患者将糖皮质激素作为一线药物治疗,其中有超过一半患者临床症状有所改善。但激素在治疗减量过程中非常容易造成复发,且长期应用激素易导致免疫力下降而增加感染风险。故目前不将糖皮质激素作为MCD患者的单一药物治疗;③对于那些有全身反应、一般情况不好、合并有多器官功能障碍的MCD患者,可考虑行单药或者联合化疗,目前较为常用的治疗方案是利妥昔单抗联合或不联合CHOP(R±CHOP)。Chronowski等[25]学者采用CHOP方案显示,接近50%MCD患者的病情较长时间可以不发生恶化。但副作用比较多;④免疫调节治疗,如沙利度胺、来那度胺等药物,Puzik等[26]研究显示沙利度胺能通过降低IL-6等炎症因子的水平而发挥抗炎作用。目前也成功报道了一些单用或者联合使用该种药物治疗MCD有效的案例,该种药物效果好,不良反应少,价格便宜,部分药物未来有望成为MCD患者的只要治疗药物;⑤还有一些其他的治疗方案,如抗病毒治疗、生物治疗(IL-6受体抗体、IL-6抗体)等,目前报道较为少见,也未被广泛应用于临床,尚缺乏大规模临床研究评估其有效性。绝大多数的UCD患者行手术切除后,预后良好,复发率低,Zhang等[6]回顾性分析了145患者的研究结果显示其5年生存率可高达97%,有少数学者报道UCD向MCD转化的案例,但病例数太少,不具有代表性。对于MCD患者,往往累及多系统,预后较差,有相关文献报道,其生存期在0~14个月[27],也有研究显示其5年死亡率高达35%[7]。
综上所述,CD是临床上发病率较低的一种疾病,尤其是腹膜后UCD更为少见,其病因和发病机制尚在探索的初级阶段,临床表现及影像学常表现出多样性,因此临床上容易误诊及漏诊,对于UCD患者,首选手术切除治疗,预后良好,可长期生存,而MCD尚缺乏统一的治疗方案,预后差,死亡率高。
猜你喜欢
影像研究与医学应用(2021年3期)2021-11-30
透析与人工器官(2020年1期)2020-11-16
透析与人工器官(2020年1期)2020-11-16
浙江医学(2020年9期)2020-07-01
浙江中西医结合杂志(2019年4期)2019-05-05
浙江医学(2019年2期)2019-01-23
实用临床医学(2018年9期)2018-12-04
中国继续医学教育(2015年1期)2016-01-06
天津医科大学学报(2015年2期)2015-12-22
哈尔滨医药(2015年2期)2015-12-01
