镍基氨分解制氢催化剂体系助催化剂研究进展
2023-03-12 09:25张凌峰樊亚娟冒辰辰伍士国顾红霞
无机盐工业 2023年3期
张凌峰,樊亚娟,冒辰辰,伍士国,顾红霞
(常州工程职业技术学院,江苏常州 213164)
在“碳达峰、碳中和”国家战略提出的背景下,氢气作为一种清洁、绿色的能源在燃料电池中的应用引起了各界的广泛关注。然而,由于氢气的体积密度和沸点都比较低,导致氢气的直接运输和储存效率较低,这对氢气的实际应用提出了巨大的挑战。因此,对储氢材料的研究和开发也是科研工作者努力的主要方向。氨气因其氢含量占17.6%(质量分数)、能量密度高达3 000 W•h/kg的特性,而被认为是储存氢气非常有前景的载体[1]。更为重要的是,氨分解制得的氢气中不含有碳氧化物,因为一氧化碳的存在极易引起燃料电池中铂电极中毒而导致其性能降低[2]。所以,相比于由碳氢化合物制备的氢气,催化氨气分解制氢是获得纯净氢气最有前景的一种方法。
目前,研究者们都致力于开发高活性的氨分解制氢催化剂。其中,贵金属催化剂如Ru、Ir、Pd及Pt-Ni都具有较高的催化活性[3-5]。然而,贵金属来源少、价格昂贵,限制了其应用。因而,研究者对非贵金属催化剂(Fe、Co、Ni、MoC、MnNx等)进行了大量研究[6-8]。其中,镍基催化剂因催化活性高、价格低和稳定性强而被认为是氨分解非常有潜力的催化剂[9-10]。催化剂的性能与载体的种类和结构特征、制备方法以及助催化剂的种类等密切相关。镍基催化剂体系中助催化剂主要有碱金属、碱土金属和稀土元素等。助催化剂的引入可以改善活性组分镍纳米颗粒的粒径分布、增强催化剂的稳定性、调节催化剂的酸碱性以及电子特性等,进而提高催化剂的性能。
最近,也有很多综述报道从不同的角度对氨分解体系进行总结[11-14],主要包括稀土氧化物、非贵金属催化剂、光催化氨分解等方面。但是,目前还没有专门针对氨分解体系助催化剂相关的总结。笔者以Ni基氨分解制氢催化剂为目标,综述了近年来助催化剂在Ni基氨分解制氢催化剂中的研究进展,详细讨论了助催化剂的作用机理以及对氨分解反应性能的影响,最后对助催化剂未来的研究方向进行了展望。
1 氨分解制氢反应机理
氨分解反应生成氢气和氮气,它是合成氨过程的逆反应。其反应式:
该反应体系是一个体积增加的吸热反应,升高温度和降低压力有利于反应向正方向进行。氨分解反应的过程包括氨分子吸附、表面反应和产物分子(H2和N2)脱附,具体的分解过程如下:
在金属催化剂上氨分解反应是一个逐步脱氢的过程。首先是氨分子在催化剂表面活性位点上吸附形成活化态的氨(步骤1)。步骤(2)~(4)则是被活化的氨进一步在催化剂表面活性位点上发生逐步解离,最终生成吸附态的N和H。步骤(5)和(6)则分别是吸附态的H和N在催化剂表面发生结合和脱附,形成氮气和目标产物氢气。该反应过程的速率控制步骤,不同的催化剂体系之间存在着很大差异。对于贵金属Ru和Rh,N—H键的裂解是反应的速控步骤,而对于非贵金属Fe、Co、Ni,它们的速控步骤则是N2的脱附[15-16]。反应过程中催化剂的活性中心与N原子之间的键能大小直接影响到速控步骤。DUAN等[17]通过密度泛函理论(DFT)研究了氨在Ni(111)晶面上的分解机理,发现N—H解离形成的N*原子位于催化剂的间隙位点上,两个N*原子结合生成N2脱附时的能垒大小决定了反应速率的快慢。所以,N2脱附成为了该反应的速率限制步骤。
通过对反应机理的研究,可以进一步从原理上来理解催化剂组成、助催化剂及载体对氨分解性能的影响。此外,将催化剂性能与反应机理相结合,还可以为催化剂的设计和开发提供理论指导。
2 助催化剂对镍基催化剂性能的影响
在Ni基氨分解制氢催化剂体系中,催化活性与活性组分Ni颗粒大小及分散性密切相关,活性中心的抗烧结能力决定了催化剂在使用过程中的热稳定性,而反应过程中的速率控制步骤(6)取决于催化剂体系的酸碱特性。为了解决影响催化剂性能发挥的限制性因素,引入助催化剂是改善催化剂性能的一种有效方法,它可以显著提高催化剂的活性和稳定性。接下来将具体讨论助催化剂对催化剂的酸碱性、活性组分的分散性及粒径控制、稳定性这3个方面的影响。
2.1 对酸碱性的影响
镍基催化剂体系中氮原子(N*)的结合—解吸过程是反应的速率控制步骤,N*与金属活性中心键能的强弱决定了其解吸的快慢。在该体系中,活性金属组分的电子态与催化剂的碱性强弱密切相关。提高催化剂碱性的最有效方式是引入碱性金属元素(如:碱金属、碱土金属以及稀土金属)。这些引入的碱性助催化剂中的电子会转移到活性金属镍上,使得镍的电负性增加,从而减弱活性中心与氮原子间的键能,促进N—N键生成和N2*解吸,最终提高反应的速率。
对于镍基氨分解制氢体系,增强催化剂碱性较为有效的方法是对载体进行杂原子掺杂,常用的杂原子主要包括碱土金属(Mg、Ca、Sr、Ba)和稀土元素(Ce、Zr)。IM等[18]通过球磨法制备了一系列碱土金属元素(Mg、Ca、Sr、Ba)修饰的氧化铝载体,再将活性组分镍负载于改性后的载体上用于催化氨分解制氢反应。通过CO2-TPD分析,发现改性后催化剂的碱性强度随着碱土金属元素周期的递增而增强(催化剂的碱性强度由小到大的顺序依次为Ni/Mg-Al-O、Ni/Ca-Al-O、Ni/Sr-Al-O、Ni/Ba-Al-O)。催化剂的碱性可以提高催化剂的活性,所以改性后的催化剂性能呈现出随着碱性的增强而提升的趋势。SATO等[19]制备了类水滑石结构的Ni-MgAl催化剂,并讨论了Mg与Al原子数比的变化对催化剂碱性和催化活性的影响。纯氧化镁体系显示出最强的碱性特征,随着Al含量的增加其碱性逐渐降低,碱性位点密度也随之降低。当Mg与Al原子数比由3∶1提升到1∶0时,氨转换频率相应地由0.14 s-1升高到0.51 s-1,说明碱性的增强对氨分解反应的性能有明显的促进作用。当然,氨的转化率并不是完全随碱性的增加而提高,当Mg与Al原子数比为6∶1时NH3转化率最高,在550 ℃低空速[3 000 mL/(h·gcat)]时可达98%,但是进一步增加Mg含量,零价Ni的分散性降低,最终降低了氨的转化率。除了氧化铝载体,在氧化锆、氧化钛体系中引入碱土金属也可以增强其碱性特征。OKURA等[20]分别在氧化钛和氧化锆载体中掺杂了Ca、Sr、Ba元素形成钙钛矿结构的催化剂,结果Sr和Ba的引入可以显著提升催化剂的性能,而Ca的促进作用则较弱。
另外,不同载体体系催化剂的催化性能差异也很明显,改性氧化锆负载的催化剂活性明显优于氧化钛体系的性能。这可能是因为锆的电负性比钛小导致其载体的碱性相对较强。对氧化锆载体的催化剂进行NH3-TPSR表征来分析氮气脱附的动力学,结果如图1所示。从图1看出,Ni/SrZrO3和Ni/BaZrO3这两个样品在120~130 ℃开始出现明显的氮气脱附峰,且峰的终止温度远低于Ni/ZrO2和Ni/CaZrO3,说明Sr和Ba改性的样品显著促进了氮气的脱附,加速了反应的控制步骤速率,最终提高了催化剂的活性。对于载体和催化剂的碱性增强方面,碱土金属的引入可以起到良好的效果,尤其是Sr和Ba两种元素。其在提高催化剂碱性的同时,还可以促进载体中的电子向活性金属传递,有利于催化剂表面氮原子的结合解吸,缩短反应的速控步骤,提高反应速率。
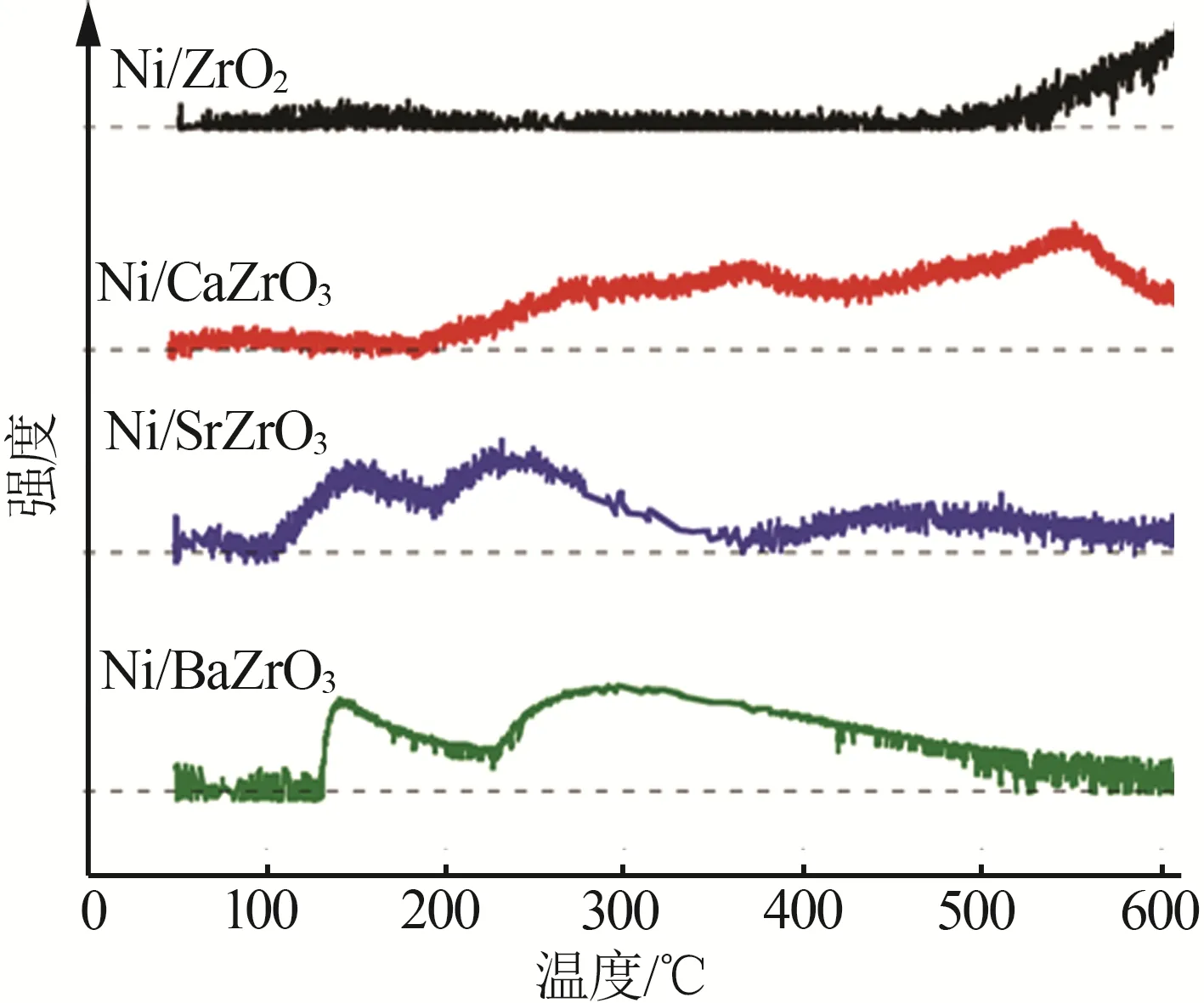
图1 Ni/ZrO2、Ni/CaZrO3、Ni/SrZrO3、Ni/BaZrO3(Ni负载质量分数为40%)催化剂NH3-TPSR测试的N2脱附曲线[20]Fig.1 N2 desorption profiles for Ni/ZrO2,Ni/CaZrO3,Ni/SrZrO3,and Ni/BaZrO3 catalysts(loading Ni with 40 wt%)obtained by NH3-TPSR measurement[20]
稀土金属氧化物掺杂到载体中也可以显著改善催化剂的酸碱特性。VACHARAPONG等[21]采用溶胶-凝胶法制备了Ce掺杂的Al2O3载体,并利用磁诱导的方法来提高Ce的分散性。负载活性组分镍后,催化剂碱性位与酸性位的比为0.99,远高于未进行Ce改性催化剂Ni/γ-Al2O3中的碱酸比(0.64)。载体中高分散的Ce可以为活性位点周围提供更高的电子密度,提高催化剂的活性。Ce与Ni之间电子转移过程:Ce4+(O2-)2-x(e-)2x+Ni=Ce4+(O2-)2-x(◇)2x+Ni(e-)2x,(◇代表CeO2表面的氧阴离子空位)[22]。路易斯碱性位点的作用机理如图2所示。从图2看出,活性位点周围高的电子密度增强了Ni与载体间的相互作用,减弱了活性组分与吸附态N的结合,这样使得N的脱附就更容易进行,加快了反应的控制步骤速率。类似地,Zr4+掺杂到氧化铝载体中也可以增强最终催化剂的碱性,提高氨分解反应的速率[23]。

图2 Ni催化剂路易斯碱性位示意图[21]Fig.2 Conceptual graphic of Lewis basic sites of Ni catalysts[21]
碱土金属元素和稀土氧化物通过合理的制备方法掺杂到载体中,这些杂原子可以向活性组分Ni提供电子,提高催化剂的碱性特征。这些增加的路易斯碱性位点通过改变Ni的电子密度,进而削弱解离的N原子与Ni中心的结合强度,促进N2的脱附,使得氨分解反应中的速率控制步骤加速进行,最终提高反应的速率。
虽然大部分情况下碱土和稀土元素的引入可以增强催化剂的碱性,但也有例外情况,例如在La2O3中掺杂MgO后碱性位点略有减少[24]。因此,需要根据载体自身的特性引入合适的助催化剂。催化剂中助催化剂引入的方式一般是先进行载体改性,再负载活性组分这两个步骤,过程相对比较复杂。另外,物理球磨法的制备工艺引入的助催化剂,其分布均匀性相对较差,最终影响催化性能。
2.2 对粒径与分散性的影响
对于负载型的镍基催化剂体系,活性金属颗粒的分散性对催化性能的影响至关重要。金属镍的高分散性有利于暴露更多的B-5活性位点[25],促进氨的催化分解。选用合适的高比表面积的载体负载活性组分,可以减小金属颗粒的粒径,进而提高其分散度。这也是提高负载金属Ni分散性常用的方法,但是可选用的载体种类不多,对粒径大小的可控性不高。然而,引入合适的助催化剂可以很好地调控活性组分的大小并改善其分散性。
ZHENG等[22]用CeO2对Al2O3载体进行掺杂改性,并讨论了不同的Ce含量对催化剂的影响。通过H2脉冲化学吸附对Ni的分散性进行了分析,适量Ce的引入提高了Ni颗粒的分散性,减小了其粒径,使得B-5活性位点增多,提高了催化剂的活性。当进一步增加Ce含量时,金属Ni的粒径会更小,但催化剂的活性却下降了。这主要是因为过多的CeO2覆盖了Ni颗粒,使得可暴露的Ni减少,导致Ni的分散度降低。VACHARAPONG等[21]进一步研究了Al2O3载体中Ce的组成和均匀性对负载的金属Ni的影响。采用磁诱导的方式提高了Ce在载体中的分散性,且负载的Ni分散性显著提高,最高可达3.15%,而没有改性的载体负载的活性组分的分散性仅为1.01%。采用透射电镜(TEM)进一步对比分析了Ce掺杂对Ni粒径的影响,Ni/γ-Al2O3催化剂中Ni的粒径为8~45 nm,而改性后载体上负载的Ni颗粒粒径降至3~15 nm。Ce修饰后的氧化铝载体对减小活性组分粒径和提高分散度具有显著的效果,进而提高了催化剂的活性。对于非负载型催化剂体系,Ce的掺杂也起到促进作用。ZHANG等[26]制备的壳核结构的Ce-NiO@SiO2催化剂,适量Ce的引入也减小了Ni的粒径,提升了催化活性。
Zr改性的Al2O3载体负载活性组分Ni也有助于提高催化剂的活性,这主要归因于改性后的载体提高了Ni的分散性,同时减小了Ni晶粒的直径,暴露出更多的活性位点,促进了氨分解反应式(2)的脱氢过程[23]。另外,适量的La掺杂到载体中也同样起到改善Ni分散性的作用,当La与Ni原子比为0.06时,金属Ni粒径由原来的3.6 nm减小到了2.8 nm,同时分散性则由25.4%提高到30.8%[27]。在CeO2-ZrO2体系(Ni/Al-Ce0.8Zr0.2O2)中,Al作为CeO2掺杂的第二组分金属,也可以提高Ni的分散性,主要原因是Al的引入增加了载体的比表面积[28]。
在Ni基氨分解制氢催化剂体系中引入适量的助催化剂(Ce、La、Zr、Al等)可以有效控制活性组分Ni的粒径大小,提高其分散度,进而促进反应的进行。然而,大部分研究都局限于描述助催化剂引入后粒径和分散度变化的现象,对机理的探究相对较少。接下来可以加深对机理的研究,为助催化剂的优选提供理论指导。
以上提及催化剂的物性与活性比较汇总于表1。
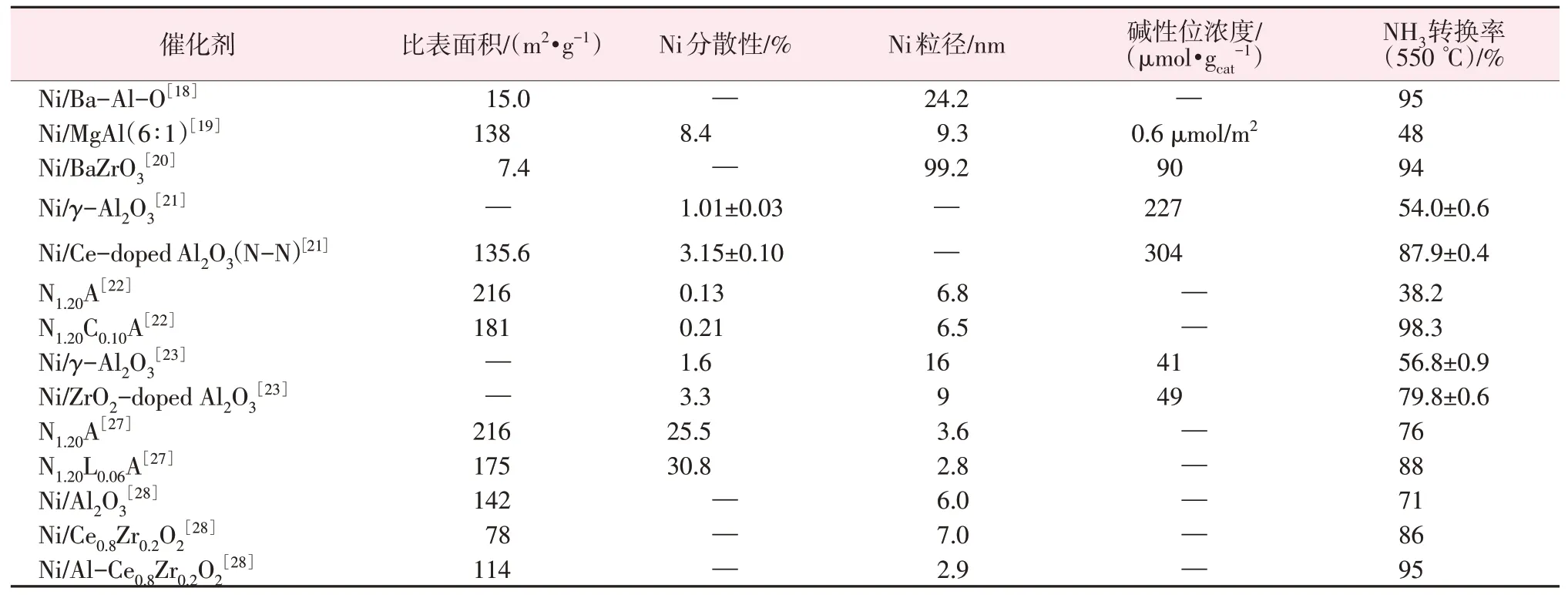
表1 催化剂的物性与活性比较Table 1 Comparison of physical properties and activity of catalysts
2.3 对稳定性的影响
除活性外,催化剂的稳定性也同样重要,尤其是氨分解制氢需要在高温条件下进行。Ni@SiO2核壳结构的催化剂可以有效抑制Ni纳米颗粒的烧结,进而提升催化剂的稳定性[26,29]。然而,这类结构的催化剂中SiO2壳层的厚度比较难控制,而且过厚的壳层会对催化活性起到负面作用[30-31]。为了提高催化剂的稳定性,引入合适的助催化剂也能达到理想的效果。
WANG等[32]研究了YSZ载体中助催化剂Ba的引入对NiO颗粒还原性的影响,H2程序升温还原(H2-TPR)分析结果如图3所示。图3中第II个还原峰归属于与载体相互作用的NiO活性组分颗粒的还原(Ni2+→Ni0),Ba的引入使得还原温度由370 ℃提高到398 ℃,说明NiO与载体之间的相互作用增强,提高了催化剂的稳定性。SIMA等[28]对比了助催化剂Al对氨分解制氢催化剂稳定性的影响,结果如图4所示。从图4看出,经过24 h稳定性测试,没有添加助催化剂的Ni/Ce0.8Zr0.2O2催化剂活性由最初NH3转换率为73%降低至68%,而Ni/Al-Ce0.8Zr0.2O2的活性在整个周期内没有表现出明显的失活迹象。这主要归结于第二助催化剂Al的引入增加了表面的氧缺陷位,进而增强了金属镍颗粒与载体间的相互作用,抑制了活性组分镍在高温下的团聚,最终提高了催化剂的稳定性。
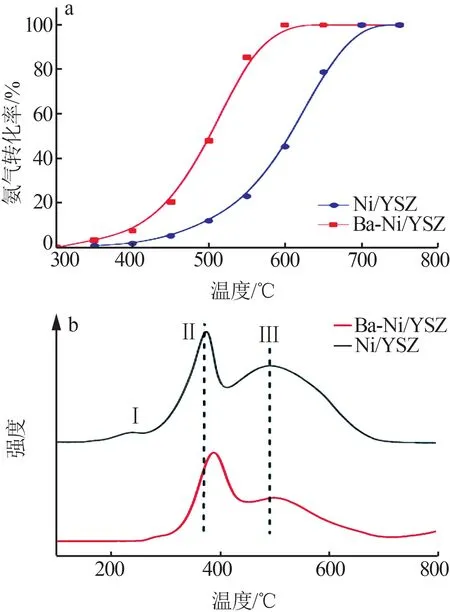
图3 Ni/YSZ和Ba-Ni/YSZ催化剂的活性(a)和H2-TPR结果(b)[32]Fig.3 Activity(a) and H2-TPR results for Ni/YSZ and Ba-Ni/YSZ catalysts(b)[32]

图4 氨气催化转换活性(a)和稳定性(b)测试结果[28]Fig.4 Test results of ammonia catalytic conversion activity(a)and stability(b)[28]
此外,合适的助催化剂还可以起到隔离的效果[22]。随着Ce与Ni物质的量比提高,α-NiO和γ-NiO所占的比例也随之升高,这主要是由于CeO2的隔离作用使得不同类型的NiO分离效果更加明显。同时,CeO2还可以抑制Ni原子的移动,阻止了粒子的团聚,增强了催化剂的稳定性。助催化剂La在Ni基催化剂体系中也被报道有类似的隔离作用[33]。
对于提高催化剂的稳定性,助催化剂的作用主要体现在两个方面:一是增强Ni与载体之间的相互作用;二是隔离金属颗粒。这两种方式都有效地抑制了活性组分的迁移团聚,最终提高了催化剂的稳定性。
3 结论与展望
在氨分解制氢催化剂体系中,Ni基催化剂由于相对低廉的价格以及较高的催化活性而展现出优越的应用前景。Ni基体系中,合适的助催化剂引入进一步提升了催化剂的性能,主要表现在增强催化剂的碱性、改善活性中心分散性以及增强稳定性等3个方面。常用的助催化剂主要有碱金属(K、Na、Cs)、碱土金属(Be、Mg、Ca、Ba)以及稀土金属(La、Ce)等,虽然这些助催化剂在改善催化剂性能方面都具有一定的促进作用,但是目前对于助催化剂的研究尚有不完善的地方。存在的主要问题:大部分的研究局限于一种或多种助催化剂的引入及催化性能测试和表征,但是对于助催化剂的作用和结构变化缺乏原子尺度的探究,对催化剂合成过程中助催化剂选用的规律指导不足;虽然助催化剂可以提升催化剂的性能,但是相对于贵金属催化剂的活性仍然存在一定的差距,需要深入探究助催化剂的作用机理并进一步改善催化活性;目前助催化剂的种类相对比较局限,在深入理解氨分解制氢微观机理的基础上,应扩大助催化剂的选用范围,开发更加高效的助催化剂。针对目前助催化剂研究存在的问题,未来的研究可以从以下几个方面来开展:1)表征方面,对于助催化剂影响的研究表征主要局限于TPR、TEM、扫描电镜(SEM)等常规方法,但是这些表征方法并不能精确地分析助催化剂的作用,需要从原子尺度以及原位的表征进行研究;2)制备方法方面,目前常用的制备方法有浸渍法、沉淀法、机械混合法等,这些制备方法很难做到原位混合,可以尝试一些原子尺度的方法进行精准调控,如化学气相沉积(CVD)法、单原子沉积法等;3)机理和应用方面,对于不同类型的助催化剂,研究其影响性能的原因并总结成一般规律,为以后催化剂助催化剂的筛选提供指导。
猜你喜欢
建材发展导向(2021年24期)2021-02-12
中国粉体技术(2021年1期)2021-01-04
上海建材(2020年12期)2020-04-13
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中国塑料(2016年4期)2016-06-27
浙江农业科学(2016年11期)2016-05-04
当代化工研究(2016年5期)2016-03-20
广州大学学报(自然科学版)(2015年4期)2015-12-23
电源技术(2015年2期)2015-08-22
电源技术(2015年11期)2015-08-22