
AP在氟化石墨烯表面分解机理的密度泛函理论研究
2023-03-11 09:52:28杨秀荣张梓涵赵子航李嘉辰马海霞
火炸药学报 2023年2期
张 驰,杨秀荣,张梓涵,赵子航,李嘉辰,马海霞
(西北大学 化工学院&西安特种能源材料重点实验室,陕西 西安 710069)
引 言
随着对导弹和火箭发动机的要求不断提高,除了改善其发动机的机械结构,提高导弹和火箭发动机的主要能量材料——复合固体推进剂的性能同样是一种可行且高效的手段。高氯酸铵(AP)作为广泛使用于复合固体推进剂配方中的一种氧化剂,在推进剂中的用量所占的质量分数为60%~90 %,其燃烧性能对复合固体推进剂的燃烧行为有重要影响[1-3]。例如,推进剂的燃烧稳定性、推力等性能会受到AP的分解活化能和高温分解温度等性质的显著影响[4-7]。大量研究表明添加剂的使用能够明显改善AP的燃烧性能[8],激发了研究者对AP分解反应机理的探索[9-17]。研究表明,在AP热分解过程中,由于分解生成的NH3分子会吸附在AP晶体表面,随着NH3吸附量的增加,会阻止AP晶体持续分解生成NH3和HClO4分子,从而阻止AP的完全分解。随着温度的升高,吸附在AP表面的NH3分子逐渐脱附,AP继续分解,直到反应完全。因此导致纯AP的热分解被分割为低温分解阶段(LTD)和高温分解阶段(HTD)[18-21]。这种热行为会导致AP基固体推进剂的点火延迟时间延长,燃烧速度较低[22-23]。因此选择合适的AP燃烧催化剂对消除阻滞期以及提高固体推进剂的性能有重要影响。采用理论计算方法研究AP在催化剂表面的反应,了解AP在催化剂表面分解的具体反应机理,可以避免实验后催化剂性能不理想而造成的物质浪费,并且能够对后续可能的实验研究起到启发和指导作用。
氟化石墨烯(FG)是石墨烯的部分或全部碳原子通过与氟原子形成共价键后生成的二维层状石墨烯衍生物。FG具有高耐热性、高导热性、自由基特性等[24-27]。因此FG作为一种石墨烯功能化改性材料逐渐受到广泛地关注以及研究,并且其在电子器件(超级电容和传感器等)、化学电源、生物材料以及催化材料等诸多领域都展现出了良好的应用前景[28-30]。含氟炭材料如聚四氟乙烯和氟化石墨等由于C—F共价键具有高强度和高能量,可以用于提高固体推进剂燃烧效率[31-32]。而FG不仅具有可调控密度的C—F共价键,同时具备二维片层结构,有望作为包覆剂用于含能材料、固体推进剂等领域[33-35]。
本研究对AP与FG间的相互作用进行了分子层面的探讨,分析了将FG用于AP燃烧催化的可行性。采用密度泛函理论方法对AP在FG表面的分解机理进行了研究。分别计算了AP的初始分解产物HClO4(PA)和NH3分子在FG表面的分解历程。对于HClO4分子,基于文献提出了两条不同的分解历程以及可能发生的基元反应,根据过渡态理论对这些基元反应进行了研究。通过对计算得到的所有基元反应的活化能以及反应热的分析,建立了HClO4分子在FG表面分解的反应网络并探讨了HClO4分解的具体分解路径。采用同样的方法研究了NH3分子的分解行为。最后,对HClO4和NH3分子间可能的相互影响进行了讨论,以推测AP整体在FG表面的分解情况。通过对AP在FG表面分解机理的分析,有助于了解催化剂促进AP分解的具体原因以及作用方式,进而为运用理论计算方法筛选出促进AP分解催化剂提供理论依据。
1 计算方法及模型
密度泛函理论经过Thomas-Fermi模型的提出、Hohenberg-Kohn理论的证明,最终通过Kohn-Sham方程的建立逐渐发展并最终进行实际的应用。DFT方法的总能量的表达式如式(1)所示:
E(ρ)=ET(ρ)+EV(ρ)+EJ(ρ)+EXC(ρ)
(1)
式中:ET、EJ、EV和EXC分别表示电子动能、库伦作用能、电子与原子核间吸引势能以及交换-相关能。伴随着泛函梯度的引入,交换-相关能变得更加精确而得到广泛的使用。
本研究所有理论计算均使用基于密度泛函理论开发的Materials Studio软件(Version 8.0)中的Dmol3模块完成[36]。采用了广义梯度近似(GGA)[37]中的Perdew-Burke-Ernzerhof (PBE)[38]交换关联势计算了电子交换关联能。采用迭代子空间普洛伊的直接反演技术(DIIS)加速系统的SCF迭代速度。根据系统的对称性,使用Monkhorst-Pack网格计算方法对布里渊区进行采样,在进行结构优化时将网格k点设置为2×2×1,而在进行性质计算时则将网格k点设置为3×3×1。采用DFT+D中的Grimme van der Waals方法对AP分子及其分解产物与氟化石墨烯表面间的分子间弱相互作用进行校正[39]。在搜索能量最低的几何结构的过程中,能量、力、位移和自洽场(SCF)的收敛标准分别设定为1.0×10-5Ha(1Ha=27.21eV),0.002Ha/Å,0.005Å和1.0×10-6Ha。根据Methfessel-Paxton方案确定了电子占用率,为0.001Ha。在研究反应机理的过程中,采用完全线性同步联合二次同步(complete Linear Synchronous Transit/Quadratic Synchronous Transit,complete LST/QST)方法对各基元反应的过渡态进行搜索,随后通过频率计算及NEB方法确定所得到的过渡态结构是属于最低能量路径(MEP)上的正确过渡态[40]。
反应活化能Ea以及反应热Er的定义如式(2)及式(3)所示:
Ea=ETS-EIS
(2)
Er=EFS-EIS
(3)
式中:EIS、ETS和EFS分别为反应初始态(initial state,IS)结构、过渡态(transition state,TS)结构以及最终态(final state,FS)结构的总能量。
吸附质在催化剂表面的吸附能(Ead)根据式(4)进行计算:
Ead=EB/surface-(Esurface+EB)
(4)
式中:EB/surface、Esurface和EB分别为吸附体系、催化剂表面以及独立的吸附质的总能量。当吸附能为负值时,表示吸附质在吸附剂上的吸附是稳定的,吸附过程为放热反应。
根据文献报道[41],在F原子含量相同的情况下,双面吸附的FG的结构比单面吸附更稳定,同时当FG的原子百分比浓度为25%时(原子比率C∶F为4∶1),FG能够稳定存在。因此本研究构建了4×4的FG超晶胞(包含32个C原子和8个F原子),为防止层间相互作用,沿z轴方向设立了15Å的真空层,氟化石墨烯表面结构如图1所示。

图1 氟化石墨烯(FG,CF0.25)晶胞结构的顶视图和侧视图Fig.1 The supercell structure of single-layer FG
由于在构建FG时,并未对晶胞进行固定,几何优化后的FG晶胞相比于洁净的石墨烯晶胞有轻微的晶胞参数变化,晶胞参数列于表1。

表1 单层氟化石墨烯以及石墨烯超胞晶胞参数Table 1 The geometric parameters of single-layer FG and pristine graphene supercell
由于F原子含量较低,F原子修饰后的石墨烯晶胞仅有轻微扩大。由于F原子在石墨烯表面的吸附,与F原子直接相连的C原子沿z轴方向向F原子的方向轻微移动,这导致石墨烯表面上C—C键的键长发生显著变化,在洁净的石墨烯超晶胞中,C—C键的键长平均为1.42Å,而在FG超晶胞中,C—C键的键长处于1.374~1.528Å范围内。根据超晶胞以及FG的对称性,FG表面共有4种不同化学环境的C原子,这4种C原子分别以罗马数字标记于图1中。
2 结果与讨论
2.1 HClO4及其分解产物在FG表面上的吸附
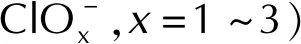
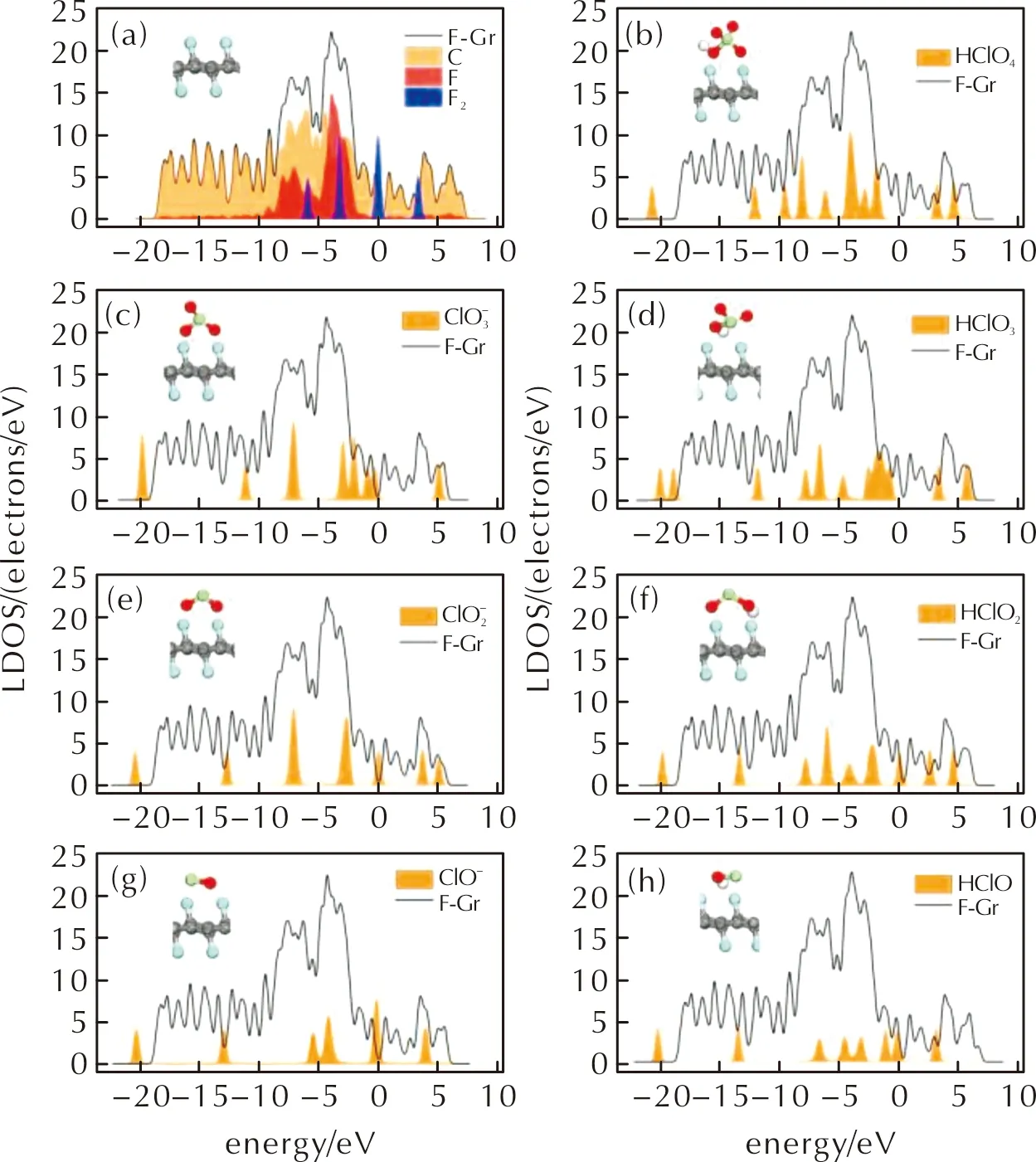
图2 (a)洁净的FG表面和(b~h)HClO4及其分解产物在FG表面上吸附结构的侧视图及其局域态密度(LDOS)图Fig.2 (a) Adsorption structures and local density of states for clean FG surface and (b—h) HClO4 as well as its decomposition products on FG surface
由图2(a)可知,随着F原子在石墨烯表面上的吸附,F原子的态密度峰发生杂化,与石墨烯上的碳原子DOS峰发生重叠,F原子的DOS峰广泛分布于C原子的分布能级-20~7eV范围内,并主要与能量在-9~-2eV能级处的C原子DOS峰重叠。F原子DOS峰与C原子DOS峰之间的全面重合,表明了F原子在石墨烯表面的化学吸附。由图2(b)可知,当HClO4吸附在FG表面时,HClO4分子的DOS峰与FG的DOS峰完全没有重叠,而吸附了HClO4分子的FG的DOS峰的数目与能级基本与洁净的FG表面的DOS峰一致,表明当HClO4分子吸附在FG表面时,两者间几乎没有发生电子的转移(计算得到的HClO4分子的mulliken电荷数为0.002e)。HClO4分子在FG表面吸附的几何结构和电子结构均表明HClO4分子的吸附为物理吸附,因此HClO4分子在FG表面的扩散及移动是相对容易发生的。如图2(c~h)所示,HClO4分子分解产生的各种中间产物在FG表面的吸附结构的LDOS图与HClO4分子的LDOS具有类似的情况,表明这些中间产物均为物理吸附。

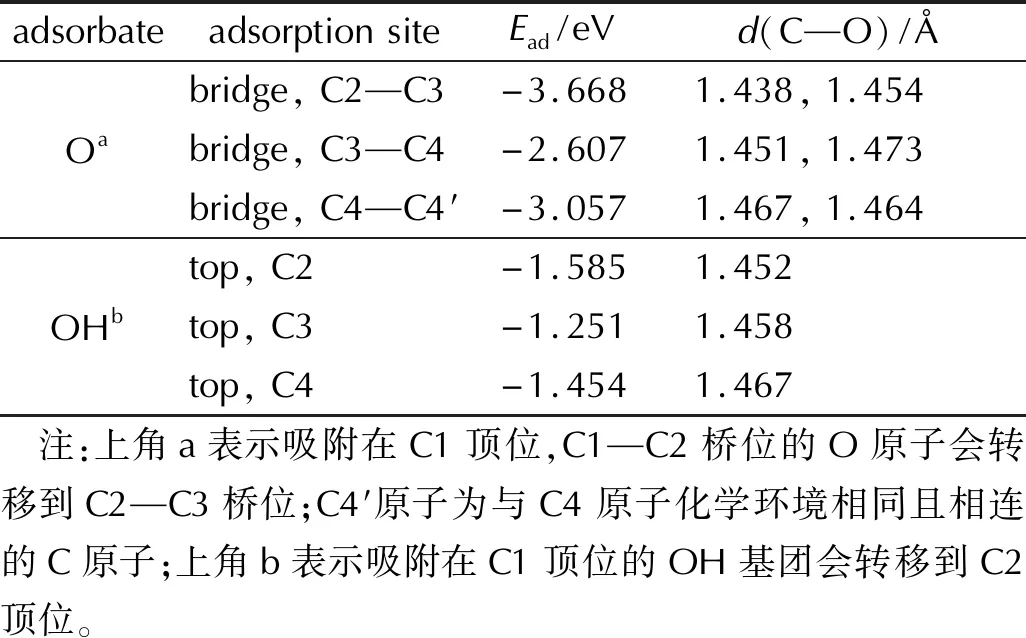
表2 氧原子及羟基基团在FG表面上的各吸附位点的吸附能以及C—O键键长Table 2 The adsorption energies and C—O bond lengths of oxygen and hydroxyl group on FG surface
当O原子吸附在FG表面时,在C原子顶位吸附的O原子会转移到C原子桥位与两个桥头C原子形成两个C—O键,吸附在不同位置的O原子经过完全优化后得到3种稳定的吸附构型,其中O原子吸附在C2—C3原子桥位时,吸附能的绝对值最大,O原子与FG间的相互作用最强,负值表示吸附后体系能量降低,吸附构型是稳定结构。因此后续计算中,O原子均吸附在C2—C3桥位。对OH基团在FG表面不同位点的吸附构型进行完全优化,部分吸附构型中吸附位点上的OH基团在优化过程中会发生转移,最终得到3种稳定的吸附结构。当OH基团吸附在C2原子顶位时,吸附能的绝对值最大,吸附结构最稳定。因此在后续计算中,OH基团均吸附在C2原子顶位。
2.2 HClO4分解的酸根路径
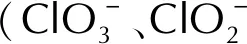
对HClO4分子在洁净的FG表面沿酸根路径进行分解的各个基元反应进行过渡态搜索,计算各反应的活化能以及反应热,确定最低能量路径(minimum energy path,MEP)。HClO4分子在FG表面的分解主要是分子中的Cl—O键的伸长以及断裂。选择HClO4分子及其分解的中间产物在FG上的吸附结构作为各基元反应的IS结构,各IS结构中的吸附质分子发生Cl—O键断裂后,解离的O或OH吸附在FG表面上的稳定结构作为FS结构。通过过渡态搜索得到的各个基元反应的过渡态结构仅有一个虚频,同时过渡态确认的结果与过渡态搜索得到的过渡态结构相一致,表明得到的过渡态结构是能量最低路径上的正确过渡态。沿酸根路径上各个基元反应的IS、TS和FS结构如图3所示,相应的反应能量曲线如图4所示。

图3 HClO4在氟化石墨烯上沿酸根路径反应的各基元反应的结构图Fig.3 Optimized structures of decomposition reactions of PA and its intermediate products along the pathway that generated oxychloride on FG surface
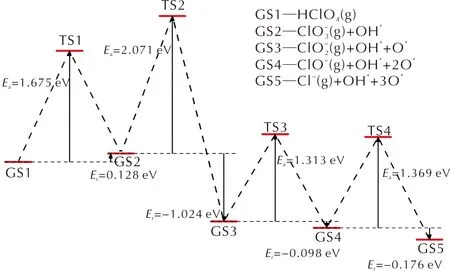
图4 HClO4在FG上沿酸根路径的各基元反应的能量曲线Fig.4 Energy profile of decomposition reactions along the chlorate route that generated oxychloride firstly

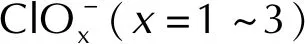
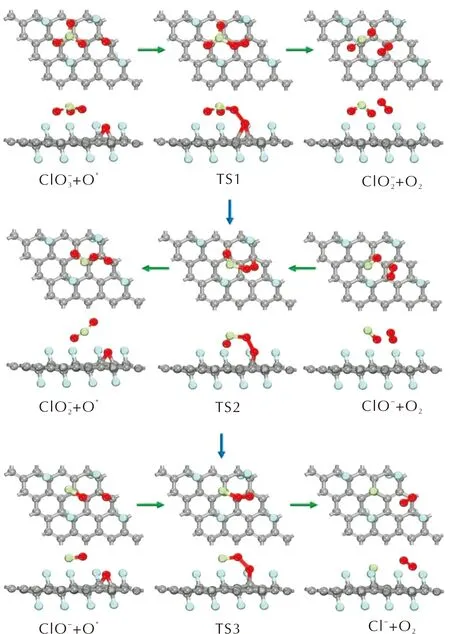
图5 HClO4分解产物在氧化FG表面反应的优化结构图Fig.5 Optimized structures of decomposition reactions of intermediate products of PA along the pathway that generated oxychloride on oxidized FG
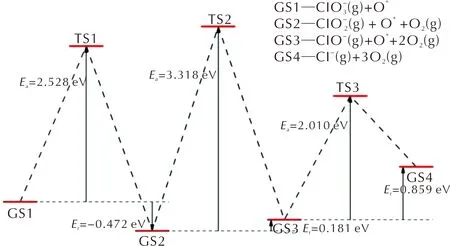
图6 HClO4分解产物在氧化FG上的基元反应的能量曲线Fig.6 Energy curves of decomposition reactions of intermediate products of PA on oxidized FG
2.3 含氧氯酸路径
催化剂的使用会改变物质分解的分解路径,研究发现,部分AP燃烧催化剂的使用会导致AP的分解产物中检测到HClO2和HClO分子的存在[43-44]。有研究者提出HClO4分子分解时会直接发生Cl—O键断裂生成HClO3分子的反应[45-46]。基于这种情况,HClO4分子除沿着酸根路径进行分解外,还可能沿下列反应路径进行分解,该反应路径记为含氧氯酸路径:
通过过渡态搜索方法对该分解路径上的各个基元反应进行了计算,得到的IS、TS和FS结构如图7所示,HClO4分子沿含氧氯酸路径分解的能量变化如图8所示。
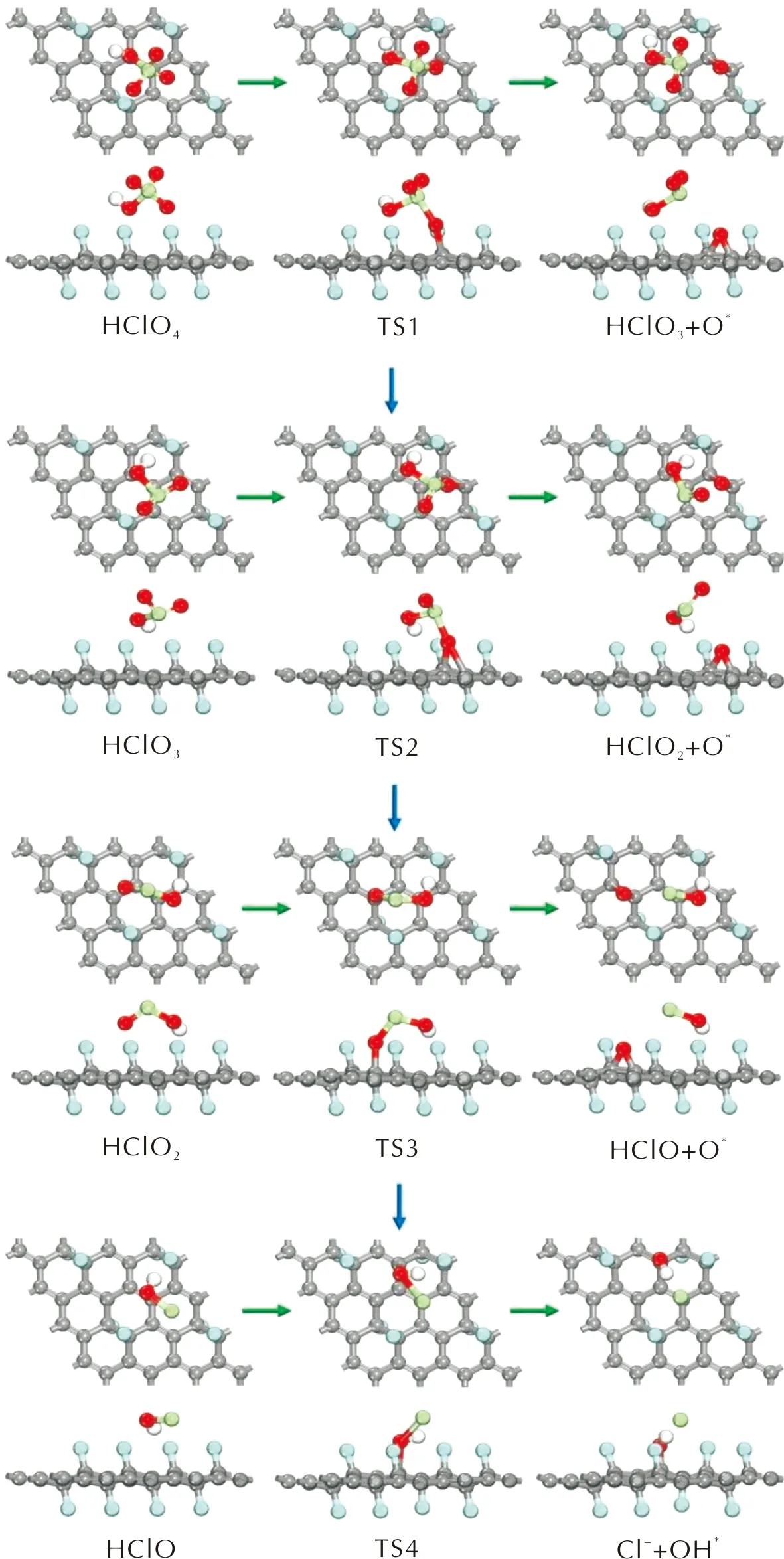
图7 HClO4分子沿含氧氯酸路径分解的各基元反应的优化结构图Fig.7 Optimized structures of decomposition reactions of PA along the oxychloric acid route
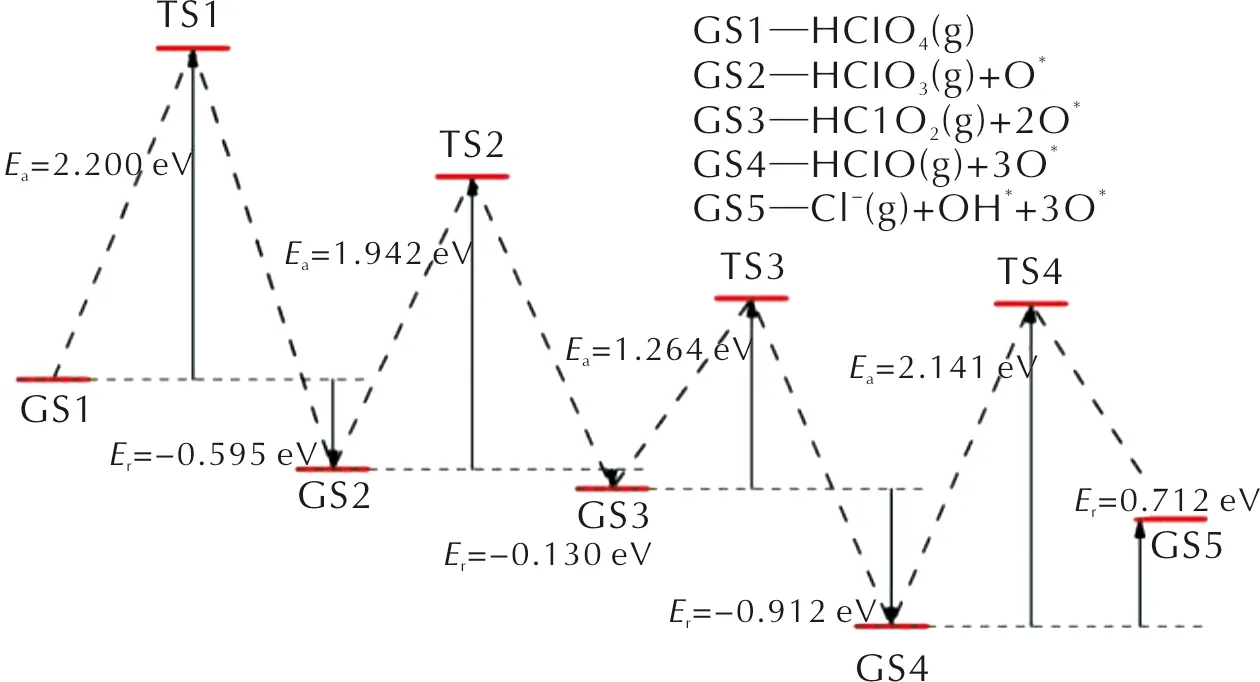
图8 HClO4分子在FG表面沿含氧氯酸路径分解的各基元反应的反应势能曲线Fig.8 Energy curves of decomposition towards elementary reactions of PA on FG surface along the oxychloric acid route
沿含氧氯酸路径,HClO4分子在FG表面首先发生Cl—O键断裂,解离出一个O原子并生成HClO3分子,解离出的O原子与表面C原子形成C—O键。该基元反应为放热反应,其活化能为2.200eV,反应热为-0.595eV。根据上述讨论,吸附在FG表面的O*原子会抑制Cl—O键的断裂反应。因此,新生成的HClO3分子在FG表面上扩散至未吸附O原子或OH基团的区域继续进行Cl—O断裂反应。HClO3分子经过Cl—O断裂反应生成HClO2分子和吸附在FG表面的O*原子,反应的活化能垒为1.942eV,反应热为-0.130eV。随后,生成的HClO2分子同样经过扩散-分解过程,通过Cl—O键的断裂生成HClO分子和O*原子。HClO2分子发生Cl—O键断裂反应时需要克服1.264eV的活化能垒,当解离出的O*原子与FG表面的C原子形成C—O键后,反应体系向外界释放出0.912eV的能量。最后,HClO分子中羟基的Cl—O键断裂生成Cl-离子和OH基团,OH基团中的O原子与表面C原子形成C—O键而吸附在FG表面,Cl-离子与OH*基团形成氢键,从而以物理吸附的方式吸附在FG表面。HClO分子的分解反应是一个吸热反应,反应的活化能和反应热分别为2.141和0.712eV。
2.4 酸根路径与含氧氯酸路径间的转换

图9 酸根路径与含氧氯酸路径上的中间产物间发生相互转化的各个基元反应的结构图Fig.9 Optimized structures of possible elementary reactions for mutual conversion between the intermediate products of acid radical path and oxychloric acid path

图10 酸根路径与含氧氯酸路径上的中间产物间发生相互转化的各个基元反应的能量曲线Fig.10 Energy profile of possible elementary reactions of intermediate products of PA on FG expect the chlorate route and oxychloric acid route
2.5 HClO4在FG表面分解反应的机理分析
从上述讨论可以发现,当HClO4分子在FG表面的进行分解时,酸根路径和含氧氯酸路径上的中间产物在一定条件下均有反应生成另一条分解路径上的其他中间产物的趋势。为了明确HClO4分子在FG表面上的具体分解路径,了解分解反应过程中反应物的消耗、中间体和产物的形成,以及各个中间产物间可能的反应关系,获得HClO4分子在FG表面上发生分解反应的反应网络图。该反应网络包含了酸根路径、含氧氯酸路径以及两条反应路径上的中间产物相互转化的各个基元反应,如图11所示。
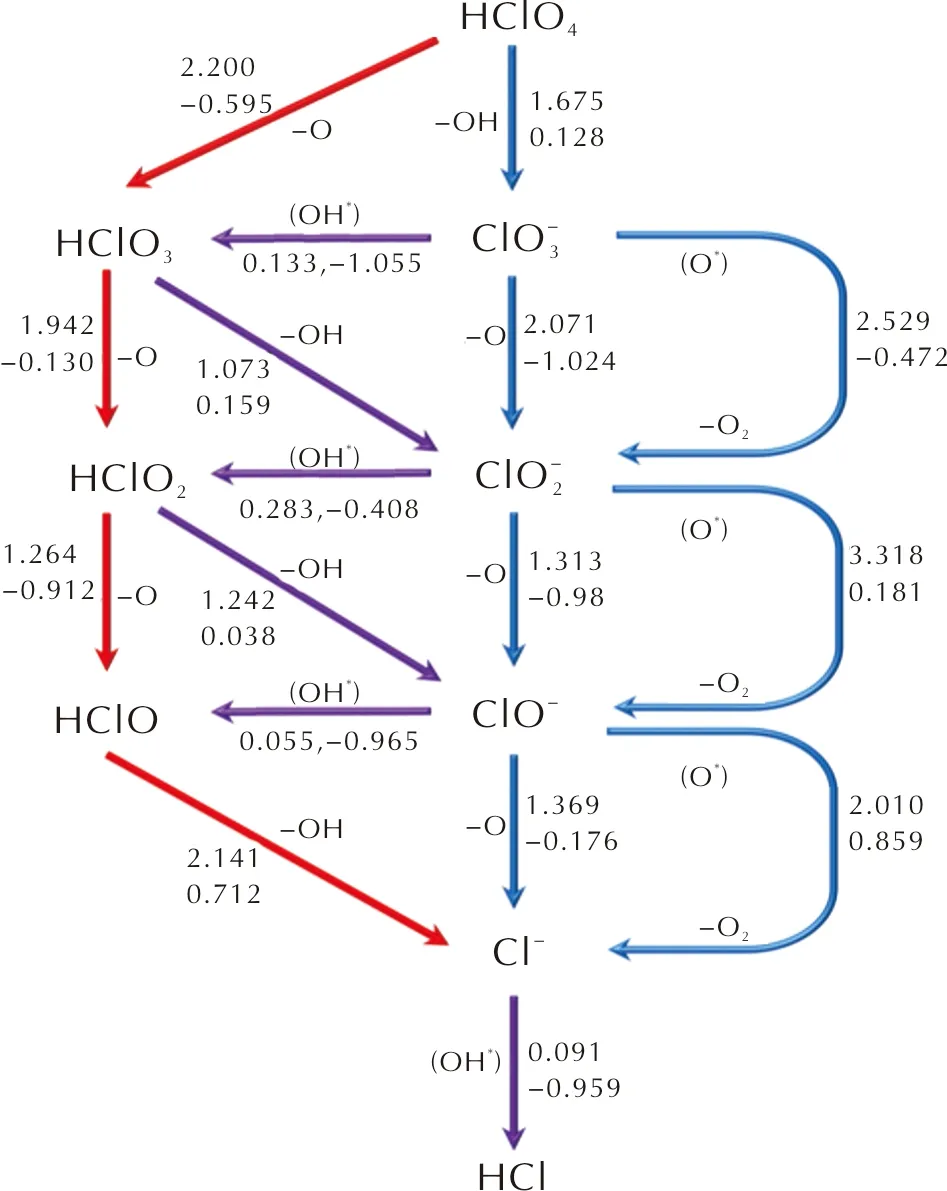
图11 HClO4在FG表面上分解反应的反应网络图Fig.11 The reaction network of the decomposition of PA on FG surface
2.6 NH3在FG表面上的连续脱氢反应
NH3分子作为AP分解的重要初级中间产物,其在FG表面的分解行为是AP分解机理的重要组成部分,同时NH3分子在FG表面的脱氢反应不仅会受到HClO4分子分解的影响,也会影响HClO4分子在FG表面的分解反应。由于HClO4分子在FG表面分解时会有大量O*原子和一定数量的OH*基团吸附在催化剂表面上,因此,不仅研究了NH3分子在洁净的FG表面的分解行为,同时也研究了其在吸附了O*原子和OH*基团的FG表面的脱氢反应。
首先研究了NH3分子在3种FG表面上的吸附情况,NH3在洁净的、预吸附了O*原子或OH*基团的FG表面的吸附结构如图12中的IS结构所示。在洁净的FG表面上,NH3分子与C4原子形成C—N键,吸附能为0.073eV。O*原子的存在会抑制NH3分子在FG表面的化学吸附,此时吸附能为0.114eV。而当OH*基团吸附在FG表面时,则会促进NH3分子在催化剂表面上的吸附,吸附能为-0.013eV。
对已经通过C—N键吸附在3种FG表面的NH3分子的脱氢反应进行过渡态搜索,各个基元反应中的IS、TS和FS结构如图12所示,反应活化能以及反应热列于表3。

表3 NH3分子在FG表面的脱氢反应的活化能Ea及反应热ErTable 3 The activation energy (Ea) and reaction heat (Er) of NH3 dehydrogenation on FG surfaces
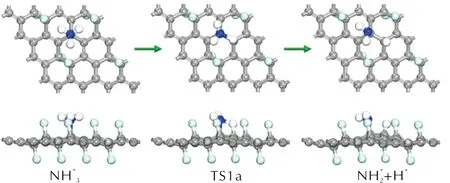
图分子在FG表面上脱氢反应的几何结构Fig.12 The optimized structures of dehydrogenation reactions of adsorbed molecule on FG surfaces
NH3分子在洁净的FG表面发生N—H键断裂,H原子与相邻的C原子形成C—H键,该基元反应是一个吸热反应,活化能和反应热分别为1.871和1.304eV。而当O*原子存在时,NH3分子与O*原子反应生成NH2和OH*基团的反应活化能降低为1.343eV,反应仍为吸热反应,但是体系需要吸收的能量降低为1.008eV。OH*基团的存在会进一步降低NH3分子在FG表面发生脱氢反应的活化能。NH3分子中的一个H原子转移至OH*基团上形成H2O分子,随后C—O键断裂,形成的H2O分子从FG表面脱附。该基元反应是一个放热反应,活化能和反应热分别为0.232和-0.673eV。O*和OH*的存在均能够活化NH3分子的N—H键,促进NH3的脱氢反应,有助于AP分解过程中产生的游离的NH3分子的分解。

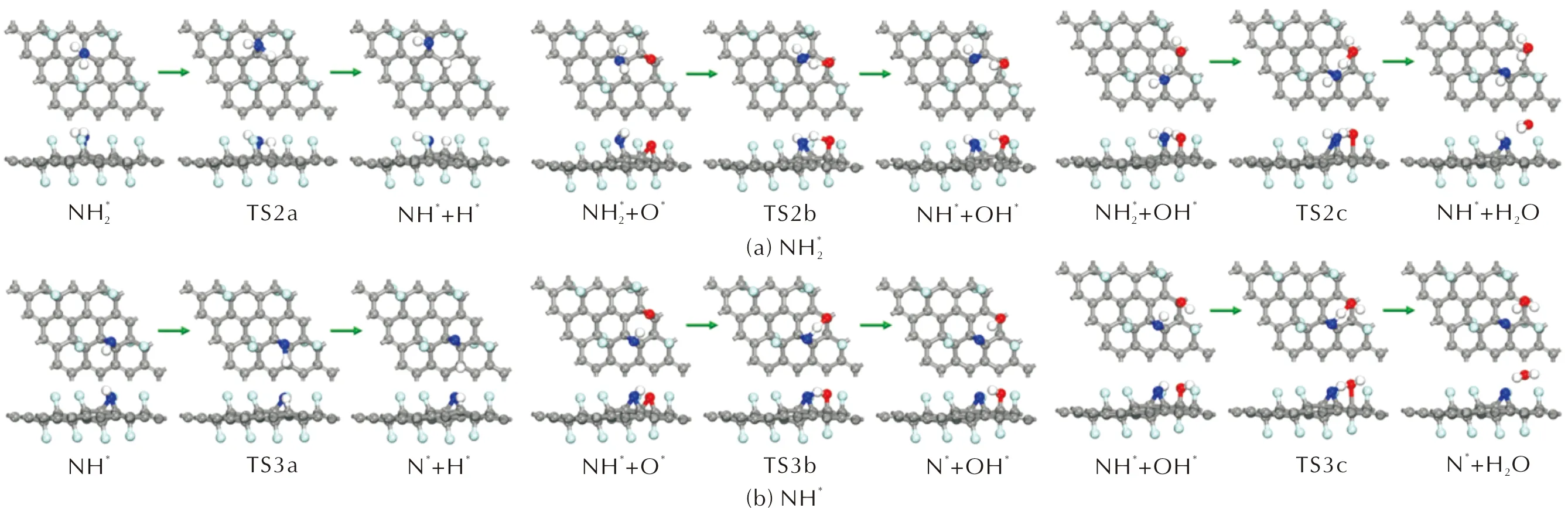
图和NH*在FG表面上的脱氢反应的几何结构Fig.13 The optimized structures of dehydrogenation reactions of and NH* on FG surfaces
不论是在洁净的FG表面还是预吸附了O*原子或OH*基团的FG表面,NH*发生N—H键断裂反应的反应历程同样与NH3是相同的。对于NH3、NH2和NH,随着脱氢反应的逐步进行,N—H键断裂反应的活化能逐渐提高。NH*在洁净的预吸附OH*的FG表面的脱氢反应活化能分别提高至2.940和1.111eV,反应热分别为2.581和0.409eV。而NH*与FG表面上吸附的O*原子反应生成N*和OH*的反应的活化能相比于NH3和NH2有所降低为1.242eV,反应热为1.195eV。
当NH3分子在洁净的FG表面进行脱氢反应时,解离的H原子除了与表面C原子形成C—H键外,还可能与F原子形成F—H键,F—H键的形成会导致C—F键的断裂,从而形成HF分子。形成的HF分子通过H原子与N*原子间形成的氢键以物理吸附的方式吸附于FG表面。对NH3在洁净的FG表面发生脱氢反应生成NHx和HF的基元反应进行过渡态搜索,反应过程中的优化结构如图14所示,各反应的活化能及反应热见上述表3。

图14 NH3在FG表面上生成HF分子的几何结构Fig.14 The optimized structures of dehydrogenation reactions of NH3 to generate HF molecules on FG surfaces
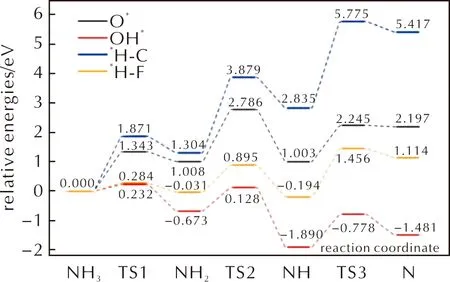
图15 NH3分子在洁净、氧化以及羟基化的FG表面上脱氢反应的能量曲线Fig.15 Energy curves of dehydrogenation reactions of NH3 on clean,oxidized,and hydroxylated FG surfaces
当NHx在洁净的FG表面与F原子反应生成HF分子时,反应的活化能仅略高于NHx与OH*基团反应的活化能,并且远低于O*存在时的活化能。因此NH3在FG表面将首先与F原子反应生成HF分子。同时由于NH3分子脱氢生成HF分子的反应活化能远低于HClO4分子分解路径上的控速步骤活化能。因此当AP在FG表面反应时,NH3分子会比HClO4分子更迅速地大量分解并生成HF分子,而不是在HClO4分子分解一定程度后才开始大量分解。这有助于AP分解过程中产生的游离NH3分子的消耗,从而防止游离的NH3吸附在AP表面。同时NH3分子的分解会降低FG表面的F原子覆盖度,导致HClO4分子在F原子覆盖度降低的FG表面进行反应。
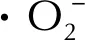
3 结 论

(2) HClO4分子分解过程中,会不断分解出O*原子以及OH*基团吸附于FG表面。OH*基团会参与到HClO4分子的分解过程中,并通过反应生成O*原子。O*原子的吸附不利于HClO4分子在FG表面的分解。
(3) AP分解产生的另一初级产物NH3分子及其脱氢产物能够与FG表面的O*原子反应生成OH*,同时其也易于与OH*反应生成H2O分子,从而降低FG表面的O*原子的覆盖度。即NH3分子在FG表面的脱氢反应促进了FG表面O*覆盖度的降低,因而有利于HClO4分子的分解。
(4) FG催化剂表面的F原子同样易于与NH3反应,从而消耗AP分解过程中产生的游离的NH3分子,防止NH3分子吸附在AP表面。AP分解过程中阻滞期的形成主要是因为AP分解生成的NH3分子在AP表面的吸附,因此FG催化剂能够有效地消除AP分解的阻滞期。
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
铜仁学院学报(2018年6期)2018-07-05 09:47:34
科学导报(2018年30期)2018-05-14 12:06:01
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
Chinese Journal of Chemical Engineering(2014年3期)2014-07-24 15:40:13
