
气相色谱-质谱法测定化妆品中斑蝥素和氮芥
2023-03-11 08:21吕琼芳彭敏梁惠明黄少宏
化学分析计量 2023年1期
吕琼芳,彭敏,梁惠明,黄少宏
(江门市药品检验所,广东江门 529000)
斑蝥素是一种天然的防御性毒素,外用具有皮肤止痒,改善局部神经营养及刺激毛根、促进毛发生长的作用[1]。但斑蝥素的毒副作用大,安全范围窄,皮肤大范围外涂易导致中毒,表现为心肌损害,心肾器官实质性损伤、中枢神经系统损害及衰竭症状[2]。
氮芥是一种生物烷化剂,有高度的化学活性,能直接作用于细胞,抑制细胞的迅速增殖,现作为广谱抗肿瘤药物应用。同时氮芥属于高毒类,是一种强起疱剂和局部刺激剂,能刺激皮肤毛发的生长。若盐酸氮芥水溶液滴到皮肤上会被迅速透皮吸收,引起大疱红肿疼痛甚至组织坏死溃疡,严重时可通过呼吸道吸收引起气道水肿和肺水肿[3]。
斑蝥素和氮芥均有促毛发生长的作用,易被非法添加在育发、防秃、洗发类产品中,但因毒副作用大,我国《化妆品安全技术规》(2015年版)规定斑蝥素和氮芥为禁用组分[4],进行严格控制,在育发、防秃、洗发类产品中均不得检出。
《化妆品安全技术规范》(2015 年版)原收载斑蝥素和氮芥的检测方法是分开的,操作不方便。规范中含量测定采用气相色谱法,样品采用三氯甲烷提取。化妆品样品的基质体系复杂[5],许多育发、防脱产品中添加了中药提取物,其生物碱成分多,用三氯甲烷提取到的杂质较多,常导致气相色谱法测定时产生很多干扰峰,对疑似阳性样品缺乏确证手段[6-9],还有需经过酸化除杂、碱性提取等繁琐的样品处理步骤,容易造成测定结果不准确。
曾有文献报道采用转相萃取-气相色谱法[10]、固相萃取-气相色谱法[7]和气相色谱-质谱联用法[11]测定斑蝥素或氮芥的含量,但少有文献将斑蝥素和氮芥同时测定,且采用气相色谱-质谱联用法定量比气相色谱法排除干扰能力更强[12]。气相色谱-质谱联用技术因其具有灵敏度高、专属性强等特点,已在化妆品定量及确证方面广泛应用[13-16],有效解决了市场上种类繁多、性状各异、成分复杂的各种育发类产品中斑蝥素、氮芥的检测问题。
国家食品药品监督管理局2019 年第12 号通告中将斑蝥素和氮芥检测方法合并,采用气相色谱-串联质谱法,但笔者在新标准实际运用中发现一些问题,针对出现的问题进行合理的优化,并对优化后方法进行方法学考察,发现优化后方法适用性更强,可行性更好。同时对影响结果准确性的关键步骤展开探讨,提出合理建议,以期为化妆品从业人员熟练运用该方法提供参考。
1 实验部分
1.1 主要仪器与试剂
气相色谱-质谱联用仪:Aglient 8890-7000D型,美国安捷伦科技有限公司。
电子分析天平:METTLER XS205DU 型,感量为0.01 mg,梅特勒托利多科技(中国)有限公司。
涡旋振荡器:Vortex3000型,维根技术(北京)有限公司。
高速冷冻离心机:CT15RT型,上海天美生化仪器设备工程有限公司。
超纯水处理系统:Milli-Q 型,德国赛多利斯集团。
盐酸氮芥标准品:批号为U 40-2008063-02,质量分数为99.69%,美国OST Research Chemicals公司。
斑蝥素标准品:批号为110783-201105,质量分数为98.6%,中国食品药品检定研究院。
三氯甲烷:HPLC级。
氯化钠、盐酸、氢氧化钠试剂:均为分析纯,广州化学试剂厂。
化妆品样品:市售。
实验用水为超纯水(符合GB/T 6682—2008 一级水要求)。
1.2 仪器工作条件
色谱柱:HP-5MS 石英毛细管柱(30 m×0.25 mm,0.25 µm,美国安捷伦科技有限公司);柱温:程序升温,初始温度为50 ℃,保持1 min,以20 ℃/min速率升至150 ℃,保持5 min,再以30 ℃/min速率升至230 ℃,保持1 min;进样口温度:230 ℃;色谱-质谱接口温度:230 ℃;载气:氦气,体积分数≥99.999%;电离方式:EI;电离能量:70 eV;测定方式:选择离子检测(SIM),氮芥定性离子为m/z106、63、120,定量离子为m/z106;斑蝥素定性离子为m/z128、96、70,定量离子为m/z128;进样体积:1 µL;进样方式:分流进样,分流比为10∶1;溶剂延迟:5 min。
1.3 实验步骤
1.3.1 标准溶液配制
精密称取斑蝥素标准品10 mg、盐酸氮芥标准品12.5 mg,分别置于10 mL 容量瓶中,用三氯甲烷溶解并定容至标线,制成斑蝥素和氮芥质量浓度均为1 mg/mL的标准储备溶液。准确移取斑蝥素和氮芥标准储备溶液各1 mL于同一10 mL容量瓶中,用三氯甲烷定容稀释制成含斑蝥素和氮芥100 µg/mL的混合标准工作溶液。分别准确移取混合标准工作溶液0.05、0.10、0.25、0.50、1.00 mL 至10 mL 容量瓶中,用三氯甲烷定容至标线,摇匀,制成系列混合标准工作溶液。
1.3.2 样品预处理
称取样品0.5 g(精确到0.001 g),置于10 mL离心管中,加饱和氯化钠水溶液2.0 mL,涡旋2 min,用0.5 mol/L 盐酸溶液或0.5 mol/L 氢氧化钠溶液将样品溶液调节至中性,精密加入三氯甲烷 5 mL,涡旋混匀2 min,于4 ℃以10 000 r/min 的转速离心10 min,取下层有机相,经 0.22 µm 微孔滤膜过滤后,滤液作为待测溶液。
1.3.3 定量方法
依次将系列混合标准工作溶液进样,分别绘制斑蝥素与氮芥的定量离子色谱峰面积-质量浓度标准曲线,其线性相关系数应大于0.99。取样品溶液进样,将斑蝥素与氮芥的色谱峰面积代入标准曲线,计算出样品中斑蝥素或氮芥的含量。
2 结果与讨论
2.1 样品处理
按照国家食品药品监督管理局2019 年第12 号通告的标准,采用5 000 r/min 离心3 min,样品尤其是膏霜类的分层不明显、不完全。然后采用4 ℃以10 000 r/min 转速离心10 min后,分层清晰。降低温度、提高转速及延长离心时间均有利于样品快速、完全分层,提取更充分,测定结果更准确。同时除杂更干净,减少了基质干扰及对仪器的污染,过滤时溶液也易于通过滤膜。
样品提取时,需要加入饱和氯化钠水溶液作为破乳剂,加入氯化钠后需要充分涡旋使基质分散,否则加入三氯甲烷提取时,乳化现象严重,使得样品的提取效率降低,影响测定结果准确性。另外氮芥在pH 7 以上的水溶液中会发生水解而失活,而pH 值对斑蝥素测定结果影响较小,故实验提取时控制pH 6 ~7,提取效果较好[1]。
2.2 色谱条件优化
改变色谱柱的程序升温速率,由标准规定的15 ℃/min 提升至20 ℃/min,其余条件保持不变时,发现分析时间缩短约2 min(见表1),此时氮芥与溶剂峰仍能有效分离。样品在分析前段有较多基质峰,继续提升升温速率基质峰易对目标峰产生干扰,故升温速率设置为20 ℃/min。针对三氯甲烷制备的样品不够稳定的情况,缩短分析时间,重现性更好,同时可以提高试验效率。
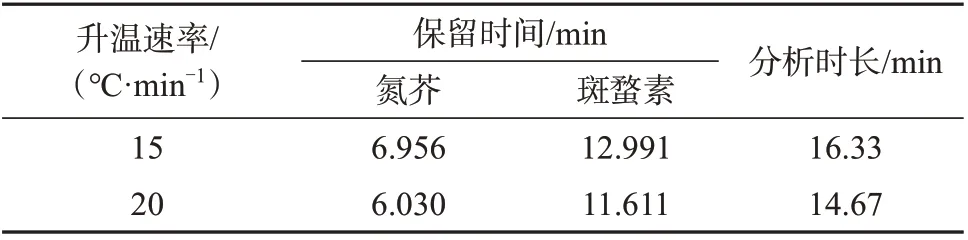
表1 不同升温速率时的分析时间
2.3 三氯甲烷试剂选择
分别采用分析纯三氯甲烷与色谱纯三氯甲烷制备样品,依次进样三氯甲烷空白溶液及用其制备的0.5 µg/mL氮芥标准品溶液,色谱图分别如图1~图6所示。
对比图1、图2并提取质谱图,确认6.304 min为氮芥峰,5.989 min为较大溶剂峰。

图1 分析纯三氯甲烷空白色谱图
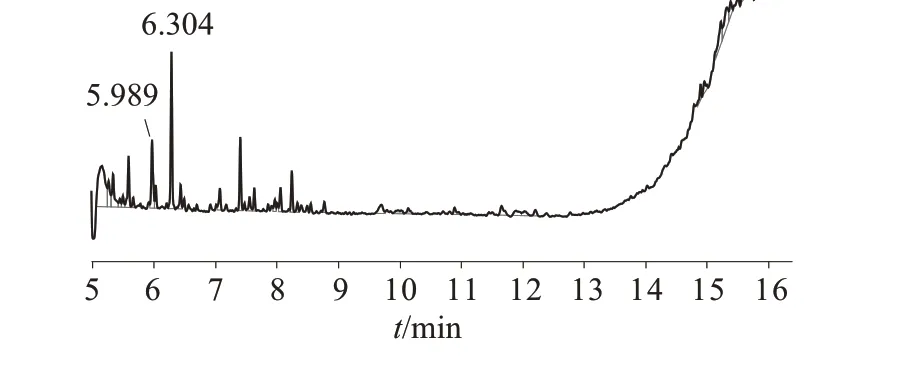
图2 分析纯三氯甲烷制备氮芥标准品色谱图
对比图3、图4,难以发现氮芥目标峰。采用色谱纯三氯甲烷制备的高浓度的氮芥定位单标和10µg/mL氮芥标准品溶液进样得图5、图6。结合图5、图6 并提取质谱图确认6.956 min 为氮芥峰,6.325 min为溶剂峰。

图3 色谱纯三氯甲烷空白色谱图

图4 色谱纯三氯甲烷制备氮芥标准品色谱图

图5 高浓度氮芥定位单标色谱图

图6 色谱纯三氯甲烷制备氮芥标准品色谱图
对比两个不同级别的三氯甲烷可见,分析纯三氯甲烷溶剂峰较色谱纯溶剂峰小很多,容易确定氮芥峰,但分析纯三氯甲烷的杂质峰较色谱纯多,对不同基质的样品干扰会更大,且长时间使用易对色谱柱及仪器系统造成污染。标准方法采用分析纯三氯甲烷,建议选用色谱纯的三氯甲烷,但尽量选择在氮芥峰保留时间附近没有溶剂峰或溶剂峰较小的三氯甲烷试剂进行试验。另外三氯甲烷应为无色透明液体,见光或久置易变黄,当发现三氯甲烷变黄色,不建议使用。
2.4 线性方程和检出限
取系列混合标准工作溶液,依次进样,进行色谱-质谱分析,以系列标准工作溶液的质量浓度为横坐标、定量离子的色谱峰面积为纵坐标,分别绘制氮芥与斑蝥素定量离子色谱峰面积-质量浓度标准曲线,计算线性方程和相关系数。
取斑蝥素和氮芥质量浓度均为 0.5 µg/mL 的混合标准工作溶液,逐级稀释,滤液进入质谱分析,测定并记录其定量离子峰峰面积。以信噪比 3∶1 对应的质量浓度作为检出限。
斑蝥素和氮芥的质量浓度线性范围、线性方程、相关系数和检出限见表2。

表 2 斑蝥素和氮芥的质量浓度线性范围、线性方程、相关系数及检出限
由表2 可知,斑蝥素、氮芥的质量浓度在0.5~10 µg/mL 范围内与各自定量离子峰峰面积线性关系良好,线性相关系数均大于0.99,方法检出限均为0.005 µg/mL。
2.5 加标回收试验
称取空白样品0.5 g(精确到0.001 g)6 份,置于10 mL 离心管中,每份分别加入0.125 mL 含斑蝥素和氮芥100 µg/mL的混合标准工作溶液。按照1.3.2步骤进行处理,制成斑蝥素和氮芥质量浓度约为2.5 µg/mL 的混合加标溶液,滤液进入质谱分析,计算回收率,试验结果见表3。
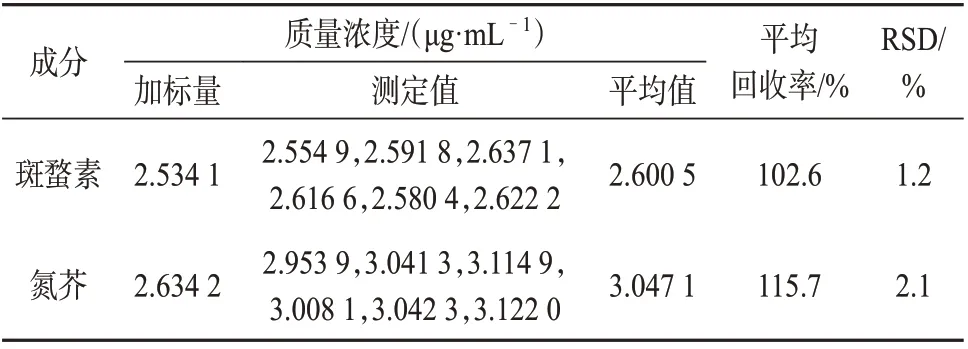
表 3 加标回收试验结果
由表3 可知,斑蝥素和氮芥的平均加标回收率分别为102.6%、115.7%,通告中标准规定斑蝥素、氮芥的加标回收率均在80.0%~120.0%之间,满足分析要求,6次平行测定结果的相对偏差分别为1.2%、2.1%,表明该方法精密度良好且准确度较高,满足分析要求。
三氯甲烷空白溶剂色谱图、混合标准品色谱图、样品色谱图以及样品加标色谱图分别如图7~图10所示。优化后的方法线性、回收率、精密度、检出限均能符合标准要求,表明优化后的方法结果准确、重现性好、适用性更强。
图7 三氯甲烷空白溶剂色谱图
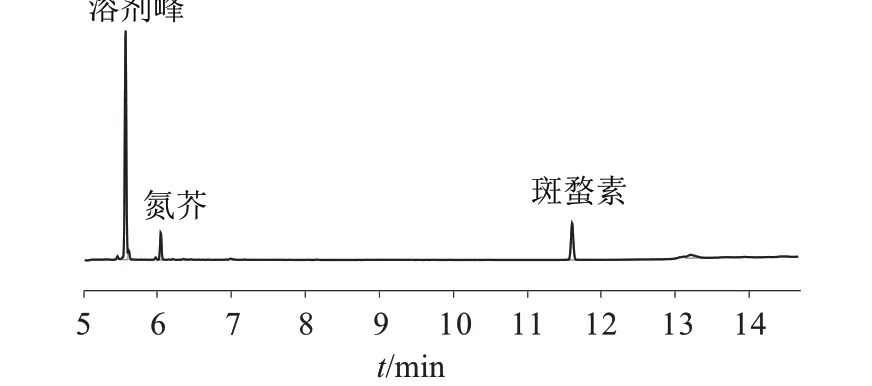
图8 混合标准品色谱图

图9 样品色谱图

图10 样品加标色谱图
2.6 标准溶液稳定性
三氯甲烷易挥发,试验中发现采用三氯甲烷溶剂制备的待测组分不够稳定。分别考察同一浓度标准品溶液(2.5 µg/mL)在临用新制、常温下和4 ℃冰箱内的稳定性,试验结果见表4。由表4 可知,常温下放置短时间内两组分色谱峰面积变化明显,且氮芥较斑蝥素色谱峰面积变化更大。为保证测定结果的准确性,建议测试溶液临用新制,或者制备后,短时间存储于4 ℃冰箱中,临进样前取出,在12 h内完成测定。

表4 同一标准品溶液不同放置条件后进样的色谱峰面积
3 结语
结合化妆品中斑蝥素和氮芥的标准检测方法在实际应用中遇到的问题,进行合理优化,优化后的方法缩短了分析时间,提高了检验效率,且方法的适用性更强,也保证了结果的准确性和重现性,适合用于育发、防秃、洗发类化妆品中斑蝥素和氮芥两种禁用组分的同时测定,为广大技术人员在应用化妆品中斑蝥素和氮芥检测方法时提供参考。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年36期)2021-03-14
科学导报(2020年75期)2020-12-21
氯碱工业(2020年6期)2020-03-01
中国化妆品(2017年12期)2017-06-27
中国化妆品(2017年12期)2017-06-27
健康女性(2017年3期)2017-04-27
当代化工研究(2016年5期)2016-03-20
特产研究(2014年4期)2014-04-10
中国环境科学(2014年4期)2014-02-02
