
白藜芦醇对阿尔茨海默病调控的研究进展
2023-03-09 13:55吴良文
食品科学 2023年3期
吴良文,张 虎,吴 桐*,陈 宁*
(武汉体育学院运动医学院,运动训练监控湖北省重点实验室,天久运动营养食品研发中心,湖北 武汉 430079)
阿尔茨海默病(Alzheimer’s disease,AD)是一种与年龄紧密相关的神经退行性疾病,其所表现出的认知与记忆功能障碍以及运动行为能力低下,可严重影响患者的生活质量。研究显示,肥胖或糖尿病人群中老年时期相比于其他人群更容易患AD,这也加大了疾病治疗的挑战[1]。据相关统计显示,全球AD患者已超过2 700万 人,俨然成为了非常严峻的社会公共卫生问题。2020年我国应对AD的费用占国民生产总值的1.47%,远超世界平均水平(1.09%),预计2030年我国应对AD年度总成本将高达5 074.9亿 美元[2-3]。
1 阿尔茨海默病的主要诱因
由于AD的病理生理学机制的复杂性,因此其组织病理学特征也成为当前研究的焦点之一,尤其是以脑组织中β-淀粉样蛋白(amyloid β-protein,Aβ)和神经元纤维缠结(neurofibrillary tangles,NFT)为主的改变。这两种病理学机制均从不同方面降低线粒体功能和突触可塑性等破坏神经元健康,进而导致AD患者进行性认知、记忆以及运动能力显著下降[4]。淀粉样蛋白前体(amyloid protein precursor,APP)经过β-位点淀粉样前体蛋白裂解酶1(Beta-site APP cleaving enzyme-1,BACE1)、β-分泌酶、γ-分泌酶切割之后,造成Aβ的产出与水解失衡,引起大量的Aβ聚集形成Aβ斑块导致大脑海马皮质细胞区域淀粉样蛋白Aβ过度堆积,最终导致神经退行性功能障碍,并引发中枢神经系统炎症反应[5]。微管蛋白Tau由于过度磷酸化而在神经元内聚合成螺旋纤维,形成NFT,破坏了微管连接的稳定,导致神经元之间的蛋白运输以及突触通信受阻[6]。虽然Aβ斑块和NFT都是AD的常见病理学特征,但是越来越多的研究表明,Aβ斑块和NFT同样预示着AD晚期[7-8]。除了蛋白质的异常聚集以外,在AD患者和实验动物模型当中也可以观察到神经元炎症、线粒体功能障碍、钙稳态紊乱以及自噬功能障碍等异常改变[9-10](图1)。

图1 阿尔茨海默病的主要诱因Fig.1 Major inducers of AD
2 白藜芦醇的生物活性
多酚是从水果、蔬菜等植物中提取的天然化合物,部分植物多酚的生物学特性对机体表现出诸多益处,如抗氧化、抗炎等。白藜芦醇(resveratrol,RSV)作为一种非黄酮类多酚,主要存在于葡萄、浆果、花生等物质中,又因其在实验研究中表现出一定的神经保护作用,因此引起众多AD相关科研工作者的关注[11]。目前,人类已经在72 种植物当中发现RSV的存在,由2~8 个RSV单元组成的相对复杂多酚,称为“白藜芦醇低聚物”,种类多达300多种。在一项针对超重老年人的记忆能力以及海马功能连接性的研究中发现,通过26 周口服RSV(200 mg/d)的治疗,老年人的记忆能力或记忆保留时间以及海马功能连接性都得到明显的提高[12]。同样,在52 周口服RSV(1 g/d)治疗中,轻度至中度AD患者与对照组相比,其简易精神状态检查评分的下降趋势得到有效的减弱[13]。除此之外,RSV可通过激活“长寿蛋白”沉默信息调节因子2相关酶1(silent mating type information regulation 2 homolog-1,Sirt1)介导的细胞自噬、神经元炎症和线粒体质量调控进而抑制Aβ生成、加速Aβ清除和衰老损伤线粒体的降解,从而达到改善AD的作用[14-15]。以上研究结果表明,RSV可能在神经退行性疾病治疗中具有潜在要用价值。不同的细胞实验以及动物实验的研究结果显示,RSV可能是一种非常有前途的防治AD的手段[11,16]。RSV作为临床AD治疗手段存在一定的缺陷。一方面是由于RSV容易与人体内葡萄糖醛酸或者硫酸盐发生反应形成RSV醛酸盐、磺基葡萄糖醛酸进而快速代谢掉,生物利用率比较低,热稳定性也相对较差;另一方面,许多早期的体外、离体和动物实验研究结果表明,RSV可通过发挥抗炎、抗氧化、促进新陈代谢的作用起到神经保护、心血管保护作用等健康益处的功效。但是随着研究的深入,RSV使用剂量和RSV补充时间的差异,可能会引起RSV剂量依赖性的毒性作用[17-18]。这主要是由于目前大多数研究都是在细胞或者动物模型上进行的,缺乏RSV治疗AD的安全性和有效性临床研究。
3 白藜芦醇对阿尔茨海默病的调控机制
目前,细胞培养实验与动物实验的结果显示,RSV可通过诱导细胞自噬、调控线粒体质量、抑制神经元炎症来改善大脑代谢,起到防治AD的作用,因此本文对相应的机制进行了梳理以便为相关研究提供参考。
3.1 白藜芦醇调控细胞自噬功能状态缓解阿尔茨海默病
随着日本科学家大隅良典因“细胞自噬”获得诺贝尔奖后,越来越多的研究发现维持正常自噬功能可能是防治众多疾病的潜在靶向机制。细胞自噬作为细胞内物质循环再利用的主要途径,通过降解体内衰老损伤的细胞器和错误折叠蛋白实现胞内“垃圾”的清除。细胞自噬分为大自噬、微自噬以及分子伴侣介导的细胞自噬3 种类型。其中,大自噬也是默认的细胞自噬方式,主要由双层膜包裹带降解底物后与溶酶体相结合形成自噬小体,最终被分解成可再次利用的氨基酸等基础底物[19]。分子伴侣介导的细胞自噬不依赖于代谢废物的囊泡递送,主要是通过伴侣蛋白热休克同源70 kDa蛋白特异性识别溶酶体膜上的五肽基序(KFERQ)靶蛋白和受体蛋白,最终由Lys-HSC70介导的底物在溶酶体中运输和降解。微自噬主要将HSC70与KFERQ蛋白结合,依靠转运必需内体分选复合物-I/II/III(endosomal sorting complex required for transport-I/II/III,ESCRT-I/II/III)实现底物转移进入溶酶体进行分解。
AD的发展过程中伴随着巨自噬和分子伴侣介导的自噬功能和水平的低下,例如,在APP转基因小鼠模型中,Beclinl蛋白表达水平下降导致细胞自噬能力减弱,显著加剧了海马组织Aβ的积聚,进而加速神经退行性变[20]。Piras等[21]发现,在原发性或继发性Tau蛋白病患者的大脑中,细胞自噬标记物微管相关蛋白1轻链3(microtubule-associated proteins 1 light chain 3,LC3)和重要自噬受体p62蛋白异常集聚,并且这两种标记物与过度磷酸化的Tau蛋白共存。在自噬-溶酶体途径已经被证明是神经元中Tau蛋白的主要降解途径的情况下,这种实验现象表明自噬-溶酶体途径的缺陷很可能有助于Tau蛋白病理学的发展。在AD的早期阶段,细胞自噬可以有效地介导Aβ、Tau的清除,但随着病情的发展,细胞自噬通路出现功能性失调,例如自噬体运输受损、自噬体与溶酶体融合不充分,以及非功能性溶酶体降解等,甚至出现自噬体与待降解物质在体内积累,同时伴有Aβ和Tau蛋白的清除效率降低而导致病情的恶化[22-23]。
RSV调控细胞自噬功能状态主要依赖于介导多种生物因子从而调控AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)的活性,抑制哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)表达[24](图2A)。RSV可通过对Sirt1的激活作用,从而实现对AMPK的调控而激活细胞自噬,降解Aβ聚集体,有效缓解AD病情的发展。Sirt1是一种重要的去乙酰化酶,参与多种生物分子代谢过程,包括神经元存活、胰岛素敏感性、葡萄糖代谢和线粒体生物发生等,在调节细胞内环境稳定等方面发挥着重要作用[25]。RSV也可通过介导酪氨酰-tRNA合成酶(tyrosyl tRNA synthetase,TyrRS)-多聚ADP核糖聚合酶1(poly (ADPribose) polymerase 1,PARP1)-Sirt1信号通路,诱导PC12细胞自噬,并且增强转运蛋白与Aβ寡聚物的结合,抑制Aβ斑块的合成,减轻Aβ引起的神经毒素[26]。RSV上调Beclin1表达,进而抑制mTOR表达,促进LC3-I向LC3-II转化,诱导溶酶体系统依赖性的细胞自噬,促使Aβ在细胞内部被降解达到改善记忆能力可能也是通过Sirt1所实现[27-28]。在APP/S1小鼠模型中,病理性细胞自噬空泡的积聚导致Aβ的聚集,这可能由于AD小鼠模型中细胞自噬通路受损致使早衰素(presenilin 1,PSEN1)的表达量升高,γ-分泌酶活力增加,最终导致Aβ合成量增加[29-30]。RSV可有效调控Sirt1活性,抑制PSEN1的表达,恢复正常的细胞自噬,抑制Aβ的产生[31]。经过RSV预处理之后,人神经母细胞瘤SH-SY5Y细胞自噬水平显著提高,可有效降低鱼藤酮的毒性作用[32]。然而,在自噬抑制剂3-甲基腺嘌呤(3-methyladenine,3-MA)的作用下,RSV的细胞保护作用被弱化[33]。此外,RSV还可以在独立于Sirt1的情况下激活AMPK,抑制mTOR表达促进可溶于细胞溶液的LC3-I转化成酯化的LC3-II,诱导细胞自噬,促进溶酶体系统对Aβ的降解[34-35]。
钙离子信号通路已经被证实可以间接调节细胞自噬的活性,钙/钙调节蛋白依赖性蛋白激酶激酶-β(calcium/calmodulin-dependent protein kinase kinasebeta,CaMMKβ)是一种在中枢神经组织中表达非常高并且受到细胞内钙离子水平调控的激酶,RSV可以通过显著增加细胞溶质钙离子水平,促使CaMMKβ被激活,与AMPK复合物结合并在Thr172位点磷酸化而抑制哺乳动物雷帕霉素靶蛋白复合体1(mammalian target of rapamycin complex 1,mTORC1),最终实现提高细胞自噬水平加速Aβ蛋白的清除[14]。目前研究表明,细胞内C a2+信号与肌醇1,4,5-三磷酸受体(inositol 1,4,5-trisphosphate receptors,IP3R)对于增加自噬通量、激活细胞自噬是必不可少的。Luyten等[36]通过向宫颈癌细胞系HeLa细胞的培养基中加入RSV(80 μmol/L、2 h)发现,RSV本身既不能影响内质网中Ca2+-ATPase活力,也不影响三磷酸肌醇(inositol triphosphate,IP3)介导的Ca2+的释放,但是RSV以严格依赖于IP3R的功能和细胞质溶质Ca2+信号的方式诱导细胞自噬激活和增强自噬通量。通过对比儿茶素与RSV对AD模型的细胞实验,虽然儿茶素在结构上与RSV有一些相似点,但是儿茶素对细胞内AMPK以及Aβ水平毫无影响,说明RSV对AMPK的激活以及其对Aβ水平的影响具有相对特异性[34]。
细胞自噬的作用可能随着AD病情的进展而发生改变,其调节的作用效果可能不是恒定的,细胞自噬的早期激活有助于神经元清除异常的蛋白质聚集物和受损的细胞器。然而,在AD晚期阶段,激活细胞自噬对于AD的作用可能较小,因为细胞自噬可能不能有效清除更成熟稳定的聚集物,甚至可能会产生负面影响。例如,在斑块和缠结形成之前,诱导细胞自噬可以改善小鼠模型的认知缺陷。但是,受损的细胞自噬通过引起突变蛋白的积累和细胞毒性,诱导蛋白质构象紊乱相关的神经退行性疾病的病因[37-38]。值得注意的是,目前针对促进细胞自噬激活的化合物的研究主要还是集中于细胞自噬诱导和自噬小体形成这一个阶段,基本上促进细胞自噬激活的化合物不会对自噬小体与溶酶体的靶向、传递、融合等相互作用产生影响。溶酶体缺陷是AD的一个标志,因而诱导细胞自噬与增强溶酶体活性将会是一种治疗AD非常有前景的策略[37]。目前,针对提高溶酶体活性的研究有限,并且主要集中在调节转录因子EB(transcription factor EB,TFEB)的活性[39]。在Tau蛋白过度磷酸化的AD小鼠模型当中,上调TFEB表达可显著降低可溶性Tau蛋白的磷酸化程度和不可溶性Tau蛋白的聚合[40-41]。因此,RSV是否能够介导TFEB活性,增强生物体内神经元的自噬——溶酶体活性还待进一步深入研究。
3.2 白藜芦醇调控线粒体质量缓解阿尔茨海默病
线粒体不仅仅只是哺乳动物细胞中通过三羧酸循环和氧化磷酸化的作用为机体提供能量的细胞器,它还在细胞凋亡、钙稳态、细胞增殖和代谢底物的产生等方面发挥了广泛的作用[42]。线粒体通常以线粒体质量控制的方式介导生物体内各种防御机制来维持生物体内正常的生理机能[43]。线粒体质量控制主要是通过调控线粒体分裂和融合的方式,维持线粒体的正常功能。线粒体的分裂主要是由包含线粒体动力相关蛋白1(dynamin-related protein 1,Drp1)的多聚体复合物介导,该复合物作用于线粒体外膜,最终实现衰老和损伤线粒体的分离。线粒体融合蛋白1/2(mitofusion 1/2,Mfn1/2)则诱导线粒体外膜融合,而视神经萎缩1(optic atrophy 1,OPA1)诱导线粒体内膜融合,从而生成功能良好的新生线粒体[44-45]。神经元是一种能耗较高、对线粒体依赖较为突出的细胞,线粒体作为ATP产生的主要细胞器,同时在调控铁稳态、Ca2+信号传导等方面也发挥着重要作用,因此神经元与线粒体质量的改变息息相关[46]。当线粒体出现质量下降时会出现活性氧(reactive oxygen species,ROS)水平的增加、Ca2+紊乱、神经传导异常、甚至出现细胞凋亡[47-48]。线粒体膜结构出现损伤引起细胞中促凋亡因子的产生,如细胞色素c(cytochrome c,Cyt c)等,激活半胱氨酸天冬氨酰蛋白酶-9(cysteine aspartyl protease-9,Caspase-9)介导的神经元凋亡,可能是诱导AD 的重要因素之一[4,49]。相关实验也证实,在线粒体氧化磷酸化功能体系受到严重损伤时,大脑皮层Aβ开始出现积聚,并且诱导ROS水平的升高而激活糖原合成酶激酶3(glycogen synthase kinase-3,GSK-3),从而促使Tau蛋白过度磷酸化,最终导致AD的特征性病理改变并加速NFT的形成[50]。此外,由于Aβ与NFT不断堆积可能会进一步破坏线粒体的功能和结构,加深AD的病情恶化[4,51]。调控线粒体质量,可能是改善神经递质传递活动、神经可塑性的有效策略。
线粒体生物发生是一个需要多种蛋白质协调调控的复杂过程,越来越多的数据表明,过氧化物增殖物激活受体γ共激活因子1α(peroxisome proliferator-activated receptor-γ coactlvator-1α,PGC-1α)与线粒体的生物发生之间的联系非常密切[52]。PGC-1α是一种具有多效性功能的转录激活因子,在线粒体生物发生过程中起着关键作用[53-54]。RSV可以直接激活Sirt1,增强Sirt1与底物的结合亲和力,从而调节PGC-1α促进线粒体生物发生,改善线粒体质量[25,55](图2B)。在一项脑出血早期损伤修复动物实验研究中,RSV可提高线粒体超氧化物歧化酶(superoxide dismutase,SOD)活力,促进PGC-1α的核易位进而提高核呼吸因子(nuclar respiratory factor,NRF)-1和线粒体转录因子A(mitochondrial transcription factor A,TFAM)水平,促使NRF-1与调节线粒体基因组转录的TFAM基因的结合位点结合,促进线粒体基因组的复制和转录[56]。Sirt1是RSV改善线粒体功能障碍发挥其神经保护功能特性的重要靶点,此外,RSV还可以通过剂量依赖的方式激活AMPK诱导PGC-1α的表达,调控线粒体质量。RSV(20 μmol/L、1 h)可刺激大鼠初级视觉皮层神经元中的AMPK和乙酰辅酶A羧化酶(acetyl CoA carboxylase,ACC)磷酸化,并且以剂量依赖方式上调PGC-1α和NRF-1的表达,提升大脑皮层神经元内的ATP水平,通过介导与调控线粒体生物发生AMPK-PGC-1α-NRF-1信号通路实现线粒体数量的增加,最终使得线粒体质量增加和功能提升[57-58]。
在模拟AD的细胞实验中,小鼠神经母瘤细胞N2a细胞在经过RSV(5 μmol/L)预处理之后,与Aβ一起进行孵育,与对照组相比,经RSV预处理过的N2a细胞,过氧化物还原蛋白和线粒体结构基因的异常表达被有效阻止,线粒体功能恢复正常[59]。RSV可以通过调控线粒体融合相关蛋白(Mfn1/2、Opa1)、线粒体裂变相关蛋白(Drp1)以及线粒体裂变1蛋白(mitochondrial fission protein 1,Fis1)的表达来调节线粒体质量水平,缓解Aβ诱导的线粒体结构和功能的损伤[49]。
由以上可知,目前RSV促进线粒体的生物发生,主要还是与其介导的AMPK激活的PGC-1α蛋白的调控有关。此外,RSV还可以通过调控线粒体融合相关蛋白以促进线粒体融合来改善线粒体的质量。因此,筛选出RSV在调控线粒体质量中的靶向蛋白有待更深入的研究。
3.3 白藜芦醇调控神经性炎症缓解阿尔茨海默病
近年来,越来越多的证据表明,Aβ异常产生、Tau蛋白过度磷酸化与神经炎症有着直接关系[60-61]。在正常生理条件下,小胶质细胞主要是通过分泌神经细胞因子,调节突触活动与炎症,维持中枢神经系统正常活动以及内环境的稳定[62]。然而,当Aβ斑块周围小胶质细胞被激活时,小胶质细胞释放大量的炎症介质如白细胞介素(interleukin,IL)-1β、干扰素-γ(interferon-γ,IFN-γ)、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、ROS和一氧化氮,致使蛋白质氧化、诱发神经元炎症,从而导致神经元变性以及未成熟血脑屏障损伤[25,62]。Chakrabarty等[63]将腺相关病毒(adeno-associated virus 2/1,AAV2/1)介导的IL-10在APP转基因小鼠大脑中表达,发现抗炎细胞因子IL-10并没有发挥积极效果,反而抑制了小胶质细胞对Aβ的吞噬作用、促进内源性载脂蛋白E(apolipoprotein E,APOE)表达。APOE通过与Aβ的聚集体结合,负向调节Aβ聚集体的清除,促使斑块进一步沉积。可见,小胶质细胞在不同情况下被活化分别体现出了保护性和有害性。
在自然衰老引起的认知障碍研究中,RSV可显著缩短24 月龄水迷宫大鼠潜伏期,并抑制TNF-α和IL-1β蛋白的表达;同时,衰老损害的空间记忆学习可能被RSV通过抑制神经炎性而改善[64]。RSV的抗炎作用可能与Sirt1抑制核因子-κB/信号转导和转录激活因子(nuclear factor-κB/signal transducers and activators of transcription,NF-κB/STAT)信号通路密切相关[65-66](图2C)。在RSV联合干细胞预处理AD小鼠实验中,RSV可显著促进AD小鼠海马神经元的形成,并伴随着海马组织Sirt1表达水平的增加[67]。当Sirt1过表达时,小鼠脑部的神经炎症减弱,最终转基因AD小鼠认知障碍得到改善[68]。值得注意的是,RSV还可以通过保护血脑屏障完整性以减少外界炎症诱导物质和炎症因子进入脑脊液,进而降低神经炎症水平。通过对AD患者血浆和脑脊液分析表明,RSV摄入表现出神经炎症水平显著降低,如炎性标志物基质金属蛋白-9(matrix metallopeptidase-9,MMP-9)在脑脊液中表达水平显著性降低[13],而MMP-9水平的降低也表明,RSV治疗可降低中枢神经系统通透性,并限制白细胞和其他炎症因子进入到大脑海马区达到保护神经元的作用。此外,RSV还可以促使Sirt1的高表达,有效抑制p53和叉头箱O(forkhead box O,FoxO)等蛋白而抑制细胞凋亡,并对神经元提供保护作用[68-69]。
目前,AD患者大脑中神经元炎症病理性变化的确切机制未完全探析清楚,以至于这些变化是该疾病的致病原因还是后果的争论悬而未决。RSV在调控小胶质细胞保护神经元的具体分子机制也尚未完全阐明,但目前RSV可调控NF-κB信号传导和丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)表达,抑制Aβ介导的小胶质细胞活化,致使多种细胞因子如TNF-α、IL-1β、IL-6、以及Cox-2的表达水平下降,抑制其介导的细胞凋亡和神经退行性变,缓解神经炎症与脑损伤,从而实现对神经元的保护。
3.4 白藜芦醇调控microRNA缓解阿尔茨海默病
miRNA是长度约22 nt左右的非蛋白质编码RNA,主要是通过与其互补的信使RNA(mRNA)靶标的3’非转录区域(3’-untranslated region,3’-UTR)结合进而抑制mRNA的翻译或促进mRNA的降解。与健康人群相比,AD患者的大脑组织中miRNA谱也发生了显著性变化[70-71]。这些改变是如何影响AD患者病情的发作和进展尚不清楚。在AD早期阶段,脑组织中异常的miRNA改变可能与机体内环境稳态与新陈代谢改变有关[72-73]。AD患者脑内Aβ的产生与沉积都伴随着不同类型miRNA的改变,如miR-17、miR-124、miR-101可以靶向调控APP与Aβ的生成,而miR-155、miR-146、miR-132可靶向调控神经炎症[74-75],值得注意的是,RSV均可对这些miRNA进行调控而实现对AD的改善或缓解作用(图2D)。
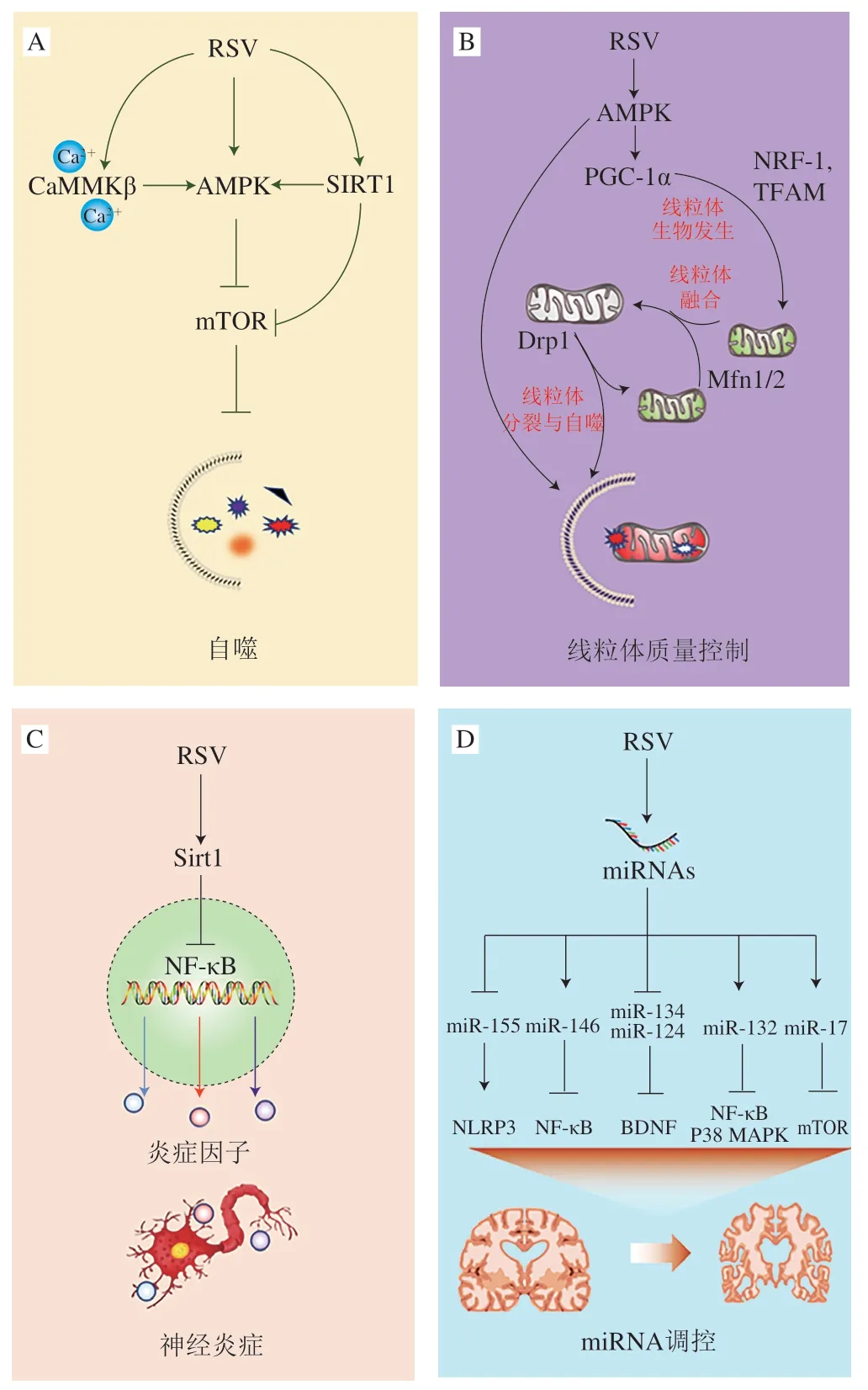
图2 RSV介导AD相关信号通路Fig.2 RSV mediates AD-related signaling pathways
miR-155表达水平在神经退行性疾病中显著增加,并且在AD发病机制中可能参与调节T细胞的分化、增殖和活化[76]。研究证实,RSV则可以通过抑制小胶质细胞中miR-155表达水平,激活AMPK和Sirt1信号通路,抑制核苷酸结合寡聚化结构域样受体蛋白3(NOD-like receptor protein 3,NLRP3)炎症小体[77]。miR-146也在小胶质细胞中大量表达,而在神经元和星形胶质细胞当中表达较少,miR-146则可直接靶向作用于IL-1受体相关激酶(IL-1 receptor-associated kinase,IRAK-1)和核转录因子受体相关激活因子6(TNF receptor associated factor 6,TRAF6)/NF-κB信号通路,而miR-146可能受到RSV的负向调控[78-79]。miR-132是许多神经元生物过程当中非常重要的调节因子,可靶向IRAK-4,负向调节IL-1β和IL-6参与调节神经系统中的先天免疫反应[80]。相关研究也显示,在PC-12细胞和大鼠模型中,RSV可通过调节miR-132的表达,阻断NF-κB和p38 MAPK信号通路,促进细胞增长和抑制炎症因子的释放,发挥抗炎特性[80]。RSV可上调miR-17以调控PTEN(phosphatase and tensin homolog deleted on chromosome ten)/磷脂酰肌醇3-激酶(phosphatidylinositol3-kinase,PI3K)/蛋白激酶B(protein kinase B,Akt)mTOR信号通路,抑制细胞凋亡,减少IL-6、IL-8和TNF-α等炎症因子的释放,继而改善细胞炎症损伤[81]。RSV-Se纳米颗粒用于100 mg/(kg·d)氯化铝(AlCl3)诱导的大鼠AD模型治疗,发现RSV-Se纳米颗粒可通过上调Sirt1的表达以抑制miR-134,从而改善神经突触可塑性[82]。在RSV介导衰老所引起认知功能下降的研究中,RSV上调脑源性神经营养因子(bain-derived neurotrophic factor,BDNF),其机制可能是通过激活miR-124和miR-134介导的环磷酸腺苷(cyclic adenosine monophosphate,cAMP)反应元件结合蛋白(cAMP-response element binding protein,CREB)-BDNF信号通路以实现衰老小鼠的学习记忆能力的改善[83]。
在健康的脑组织中,miRNA可以通过对神经元和免疫信号通路中的靶标进行调控维持体内环境的平衡。目前,越来越多的miRNA已经被鉴定出与AD患者的病理性变化有密切联系,而调控miRNA可能成为防治AD的重要干预手段,其中类似于RSV的天然产物值得进一步挖掘。
4 结语
AD在老年人群中已经逐渐成为常见高发疾病,细胞自噬缺陷、神经元炎症与线粒体质量低下被证实是AD的主要诱因,而RSV的干预则可通过调节ROS产生、抑制神经元炎症、促进细胞自噬状态优化和调控线粒体质量来改善脑组织代谢而缓解AD或抑制其病情恶化。但是,针对AD患者人群的治疗,RSV的最佳给药剂量和给药途径目前尚未完全明确探析。植物多酚的低生物利用率问题也逐渐成为人们所关注的重点,RSV单体或者类似物,是否可以通过与运动、药物等协同方式促进机体对RSV的吸收,加强血脑屏障对其的通透性,提升RSV的生物利用率,提升对AD的治疗效果有待进一步研究。此外,随着纳米、基因与生物工程技术的成熟,通过对RSV单体或者衍生物进行修饰、RSV纳米胶囊等结合日常生活方式调节和运动干预是否可以起到防治AD的作用值得进一步探索。目前研究表明,RSV主要是通过激活Sirt1、调控miRNA介导的CREB-BDNF通路,下调NF-κB的水平,诱导细胞自噬激活等机制发挥其神经元的保护作用,鉴于RSV在AD人群中的研究有限,因此,其在神经退行性疾病方面的治疗效果还需要开展临床研究获取更多的数据支持。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
生物化工(2021年2期)2021-01-19
生物化工(2020年1期)2020-02-17
读与写(2019年35期)2019-11-05
现代装饰(2018年5期)2018-05-26
现代职业教育·高职高专(2018年7期)2018-05-14
中国生化药物杂志(2015年4期)2015-07-07
弹箭与制导学报(2015年1期)2015-03-11
