
Assessment of compatibility of rhIGF-1/rhIGFBP-3 with neonatal intravenous medications
2023-03-01 05:42NazilaSalamatMillerMarkTurnerAmeyBandekarNitinDixitEmilyJochimBarryMangumChristopherMcPhersonSriniTenjarlaSukhjeetSinghYouSeokHwangNormanBarton
World Journal of Pediatrics 2023年1期
Nazila Salamat-Miller · Mark A. Turner · Amey Bandekar · Nitin Dixit · Emily Jochim · Barry Mangum ·Christopher McPherson · Srini Tenjarla · Sukhjeet Singh · You Seok Hwang · Norman Barton
Abstract Background Recombinant human (rh)IGF-1/IGFBP-3 protein complex,administered as a continuous intravenous infusion in preterm infants,is being studied for the prevention of complications of prematurity.Methods We conducted in vitro studies to evaluate the physical and chemical compatibility of rhIGF-1/IGFBP-3 with medications routinely administered to preterm neonates.In vitro mixing of rhIGF-1/IGFBP-3 drug product with smallmolecule test medications plus corresponding controls was performed.Physical compatibility was defined as no color change,precipitation,turbidity,gas evolution,no clinically relevant change in pH/osmolality or loss in medication content.Chemical compatibility of small molecules was assessed using liquid chromatography (e.g.,reverse-phase HPLC and ion chromatography),with incompatibility defined as loss of concentration of ≥ 10%.A risk evaluation was conducted for each medication based on in vitro compatibility data and potential for chemical modification.Results In vitro physical compatibility was established for 11/19 medications: caffeine citrate,fentanyl,fluconazole,gentamicin,insulin,intravenous fat emulsion,midazolam,morphine sulfate,custom-mixed parenteral nutrition solution (with/without electrolytes),parenteral nutrition solution+intravenous fat emulsion,and vancomycin (dosed from a 5 mg/mL solution),but not for 8/19 medications: amikacin,ampicillin,dopamine,dobutamine,furosemide,meropenem,norepinephrine,and penicillin G,largely owing to changes in pH after mixing.Small-molecule compatibility was unaffected post-mixing,with no loss of small-molecule content.For physically compatible medications,risk analyses confirmed low probability and severity of a risk event.Conclusion Co-administration of rhIGF-1/rhIGFBP-3 drug product with various medications was assessed by in vitro studies using case-by-case risk analyses to determine the suitability of the products for co-administration.
Keywords Chemical compatibility · Intravenous · Neonatal · Physical compatibility · rhIGF-1/rhIGFBP-3
Introduction
The rhIGF-1/rhIGFBP-3 drug product is the recombinant human (rh) version of the naturally occurring protein complex of insulin-like growth factor-1 (IGF-1) and its most abundant binding protein,insulin-like growth factor binding protein-3(IGFBP-3).The product is currently under investigation for the prevention of complications of prematurity.Fetal IGF-1 concentration increases during the later stages of pregnancy and is important in the growth and development of the fetus [1].For preterm infants,IGF-1 levels are lower during the first weeks of life than in fetuses of the same gestational age in utero [1].In addition,lower levels of IGF-1 in extremely preterm infants have been associated with retinopathy of prematurity,bronchopulmonary dysplasia,and impaired neurodevelopment [1–7].
A 2019 phase 2 trial evaluated the use of rhIGF-1/rhIGFBP-3,formulated at 50 μg/mL isotonic acetate-based solution,at pH 5.5,with minor amounts of a surfactant,for the prevention of complications in extremely preterm infants (born at <28 weeks’ gestational age) [8].Although the trial’s primary endpoint of reduction in severity of retinopathy of prematurity was not met,substantial reductions were reported in the incidence of severe bronchopulmonary dysplasia and intraventricular hemorrhage.If the findings from the phase 2 trial are replicated in larger studies,there is a potential for rhIGF-1/rhIGFBP-3 to have an important impact on the clinical burden associated with preterm birth.
A previous pharmacokinetic modeling study predicted an rhIGF-1/rhIGFBP-3 dosing regimen of ≥ 250 μg/kg/24 h via continuous intravenous (IV) infusion to maintain serum IGF-1 levels in the physiological intrauterine range in extremely preterm infants [9].However,IV access can be a challenge in preterm infants in the neonatal intensive care unit (NICU) because they typically require multiple IV therapies,including different classes of medications and parenteral nutrition (PN).Assurance of the compatibility of rhIGF-1/rhIGFBP-3 with other continuously infused medications is therefore of utmost importance.In the present study,a risk assessment strategy was designed to assess the feasibility and safety of co-administering rhIGF-1/rhIGFBP-3 with medications routinely administered IV in the NICU.This risk assessment strategy was based on the in vitro testing of physical and chemical compatibility and the theoretical potential for chemical incompatibility.Herein,we report the risk assessment methodology and findings for the medications evaluated.
Methods
Test medications
Test medications were selected on the basis of clinical priority (i.e.,medications frequently administered to neonates),as identified by clinical experts and from investigative sites for the phase 2 trial [8].The 19 medications included in this study were amikacin,ampicillin,caffeine citrate,dobutamine,dopamine,fentanyl citrate,fluconazole,furosemide,gentamicin,insulin,intravenous fat emulsion,meropenem,midazolam,morphine sulfate,norepinephrine bitartrate,penicillin G,custom-mixed PN solution (with and without electrolytes),PN solution+intravenous fat emulsion,and vancomycin (Supplementary Table 1 in the Supplementary Information).Most of the test medications were small-molecule drugs,whereas rhIGF-1/rhIGFBP-3(Takeda,Lexington,MA,USA) is a recombinant protein.
Risk assessment design and overview
The risk assessment methodology was developed by a crossfunctional team comprising clinicians,neonatal pharmacists,and representatives from the study sponsor’s product development departments for small molecules,biologics,clinical,and clinical operations.The risk assessment was composed of three consecutive stages (Fig. 1): (1) in vitro testing to determine the physical and chemical compatibility of rhIGF-1/rhIGFBP-3 with other medications (small molecules only);(2) a risk evaluation for each of the test medications,taking into account the known theoretical potential for chemical modifications,proximity to the isoelectric point of the protein when not in the mixture (based on pH value and probability of chemical modification),and the clinical co-infusion history (including co-administration with insulin [10],which shares a large homology with rhIGF-1);and(3) risk planning,based on an assessment of low,medium,or high risk of incompatibility.

Fig.1 Risk assessment design. RP-HPLC reversed-phase high-performance liquid chromatography,SEC-HPLC size-exclusion high-performance liquid chromatography,USP United States Pharmacopeia.Green box denotes compendia and USP monograph assays
Mixing protocols
A mixing model was developed whereby rhIGF-1/rhIGFBP-3 and the test medication were mixed at one or more representative clinical doses.For all studies,the mixing calculations were performed with appropriate normalizations (time and neonates’ weights) to devise a volumebased scheme.For these calculations,a dose of 250 μg/kg/24 h (the dose used in the phase 2 trial) [8,9] and at least two bracketing doses of the small-molecule medication were used (Table 1).For each study,multiple controls were designed to represent post-mixing concentrations and matrices of either rhIGF-1/rhIGFBP-3 or the test medication.The controls were created by diluting the rhIGF-1/rhlGFBP-3 drug product either with its own formulation buffer (to create a concentration control) or with the matrix of the small molecule to study the effect of change in the absence of any small molecule.
Where applicable,the mixing duration was based on the calculated average infusion rate of rhIGF-1/rhIGFBP-3 (e.g.,250 μg/kg/24 h dose normalized for a 0.5 kg neonate and the target rhIGF-1/rhIGFBP-3 protein concentration of 50 μg/mL) with each test medication at the highest dose (normalized for the same weight) and an estimated volume for an umbilical catheter (Table 1).In all studies,periodic sampling of the mixture and control solutions was performed [11].Additionally,longer mixing durations were considered as worst-case scenarios (e.g.,interruptions could occur in clinical practice,extending the duration of the infusion) and to observe the continuation of any observed phenomena that occurred at the onset of mixing.
Two administration scenarios were assumed for the mixing duration calculations,where applicable.The first scenario was when each of the two medications was administrated via a pump,where the average of the two flow rates at the highest small-molecule dose was selected.In the second scenario,only rhIGF-1/rhIGFBP-3 was pumped,while the test medication was infused over a relatively short time(~ 15–30 min).For these scenarios and any other medications that were not pumped or infused,periodic observations and sampling occurred at selected time points (Table 1).
Physical compatibility
Physical compatibility assays were compared for test samples and corresponding control solutions.In line with the existing literature [10,12–16],we used the following methods,or modified versions thereof,to assess the physical compatibility of rhIGF-1/rhIGFBP-3 with the co-infused drugs: visual observation (United States Pharmacopeia [USP] <790 >),optical density at 320 nm (USP <851 >and <857 >),pH measurements (USP <791 >),and osmolality (USP <785 >)at room temperature.
The aim of visual observation was to determine the presence of any precipitation,visible particulates,and flocculent matter,as well as any color change (compared with water)and/or gas formation,which are potential indicators of chemical modification(s).
All vials for physical compatibility testing were examined under the same lighting conditions: against a white and black background using both fluorescent light and Tyndall light (Spectralight III,Macbeth/X-Rite,Grand Rapids,MI,USA or MIH-DX,Bosch/Eisai Machinery,Waiblingen,Germany).Mixture and control samples were analyzed for appearance post mixing at specified time points (Table 1).Optical density measurements at 320 nm were carried out using an ultraviolet–visible spectrophotometer (SpectraMax M5,Molecular Devices,San Jose,CA,USA) for the detection of turbidity,an indicator of submicroscopic protein aggregation.Measurements were performed in triplicate using a 1 cm path-length quartz cuvette for each sample at each time point,and the average of these was recorded.pH values were recorded for each solution in triplicate using a calibrated pH meter (Model 215,Denver Instrument,Bohemia,NY,USA;Fischer Scientific Accumet XL150,Pittsburgh,PA,USA),and the average was reported.Osmolality changes post mixing at room temperature were recorded using a calibrated osmometer (Model 3250,Advanced Instruments,Norwood,MA,USA).Triplicate readings were ascertained,and the average was recorded.
Test medications were considered physically compatible with rhIGF-1/rhIGFBP-3 if there was no observed change in color,precipitation,turbidity,or gas evolution,or if there was no clinically relevant change in osmolality or pH.
Any change ± ~ 0.3 pH of the mixtures from that of the rhIGF-1/rhIGFBP-3 drug product control (pH 5.5) was considered a change that could impact rhIGF-1/rhIGFBP-3 quality,in which case the small-molecule medication would be considered not compatible and would require further protein-specific data for evaluation.The considered range was based on the control and release specification of rhIGF-1/rhIGFBP-3 (5.5 ± 0.3) and a priori knowledge of the potential degradation and known stability of the rhIGF-1/rhIGFBP-3 drug product.The changes in osmolality values were considered using a less stringent criterion owing to existing clinical practices and in consideration of release specifications of rhIGF-1/rhIGFBP-3 (300 ± 30 mOsmol/kg) [17].
Small-molecule chemical compatibility
The concentration of small-molecule test medications post mixing was assessed using either reversed-phase high-performance liquid chromatography (RP-HPLC),with ultraviolet (UV) detection,or ion chromatography with electrochemical detection at the last specified time point(s) (Table 1) (USP monographs or modified versions).For example,while a RP-HPLC–UV methodwas used for the detection of majority of molecules,for some,such as Amikacin and Gentamicin,a modified version of the USP ion chromatography assay with an electrochemical detection was used.For each medication,a qualification of the USP methods was conducted to ensure specificity,linearity,repeatability,and accuracy of the method.Example chromatograms for one small molecule(Gentamicin) are presented in the Supplementary information.Chemical incompatibility was considered to be a loss of the small-molecule content of~ 10% or more over the defined testing period.Small-molecule analysis was not possible for PN or lipids owing to the complex nature of such mixtures.
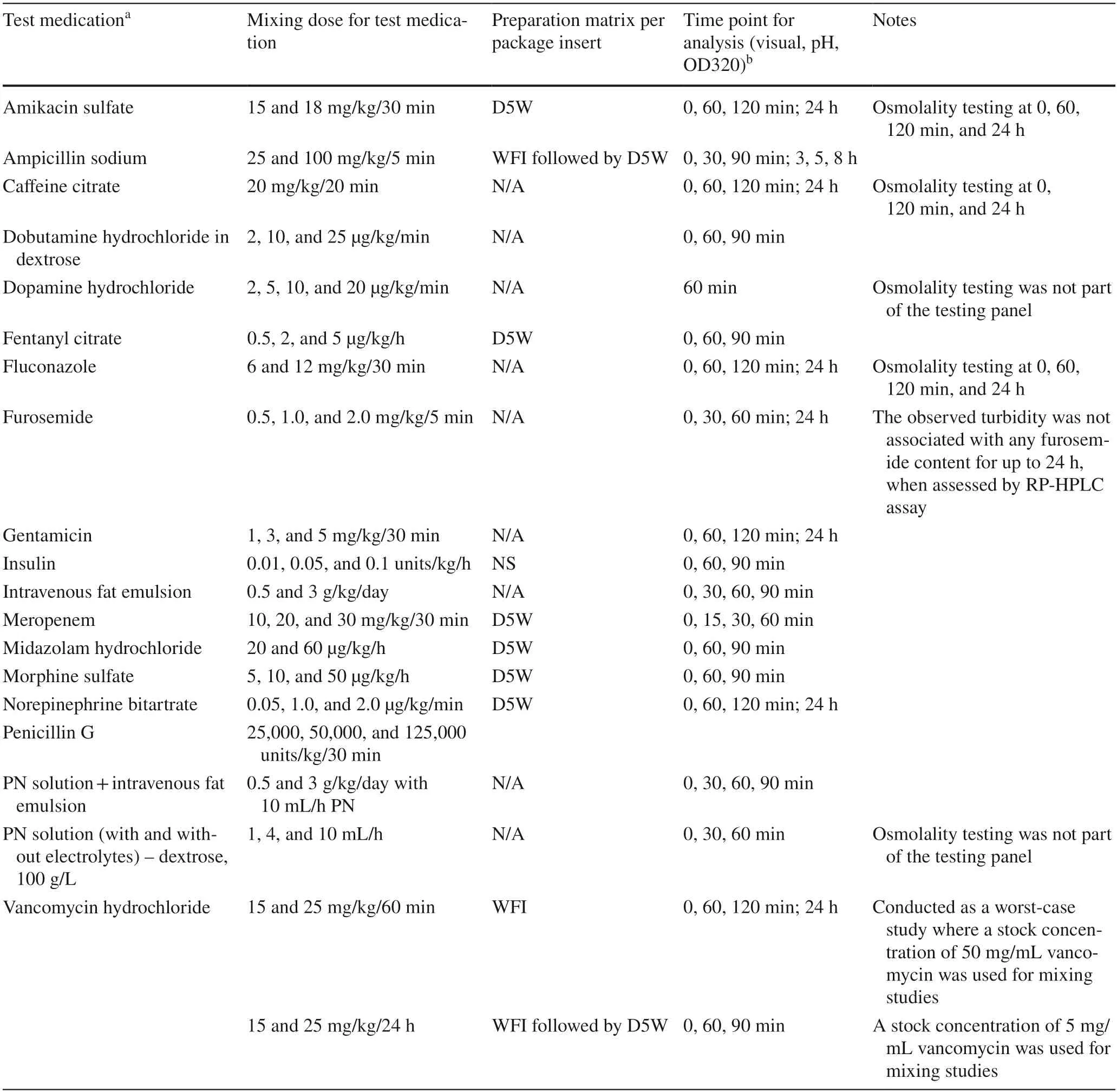
Table 1 rhIGF-1/rhIGFBP-3 drug product and small-molecule compatibility studies
rhIGF-1/rhIGFBP-3 chemical compatibility
Sensitive mass spectrometry-based protein-specific methodologies have been developed by Takeda to assess the chemical compatibility of the rhIGF-1/rhIGFBP-3 drug product.The development of protein-specific methodologies is reported separately [18].
Risk evaluation and risk planning
A comprehensive risk evaluation was completed for medications where in vitro (non)compatibility was indicated.(SeeRisk assessment design and overviewinMethodsfor a description of the risk evaluation).A risk event was defined as “rhIGF-1/rhIGFBP-3 is not compatible with the co-infused drug over the duration and condition of the simulated mixing studies”.
The risk evaluation was performed for each co-infused test medication to determine the probability and severity of a risk occurrence.Probability was defined as the likelihood of an effect on safety,efficacy,or quality;severity was defined as the severity of the impact should the risk event occur (Table 2).On the basis of the level of probability and severity (low,medium,or high),a risk planning strategy was developed for each medication (Table 3).The cross-functional team of subject matter experts performed the final assessments and endorsed the clinical recommendations.
Results
In vitro physical compatibility
Of the 19 medications tested,physical compatibility was established for rhIGF-1/rhIGFBP-3 with caffeine citrate,fentanyl,fluconazole,gentamicin,insulin,intravenous fat emulsion,midazolam,morphine sulfate,PN solution+intravenous fat emulsion,PN solution (with and without electrolytes),and vancomycin (when dosed from a 5 mg/mL solution) (Table 4).The following medications were considered incompatible with rhIGF-1: amikacin,ampicillin,dobutamine,dopamine,furosemide,meropenem,norepinephrine,penicillin G,and vancomycin (when dosed from a 50 mg/mL solution).

Table 2 Risk assessment definition
Small-molecule chemical compatibility
Small-molecule compatibility was not affected post mixing for the medications tested.No loss of small-molecule content was observed for any of the medications tested in the mixture and corresponding controls (Table 1).

Table 3 Risk prioritization grid with associated risk planning
Risk evaluation and risk planning
Risk evaluations were completed for all small-molecule test medications (except furosemide,where the studied mixture became turbid within~ 30 min and clearly indicated incompatibility with the rhIGF-1/rhIGFBP-3 drug product).Where in vitro physical compatibility was confirmed,the subsequent risk evaluations confirmed a low probability and severity of an event within the context of in-use conditions.The risk of interaction or chemical modification,based on pH values,also was considered to be low for those medications showing in vitro compatibility.
Given the structural similarity between insulin and IGF-1[19],medication compatibility with insulin was given significant consideration when evaluating rhIGF-1/rhIGFBP-3 compatibility with tested medications.For example,compatibility with insulin has been established for midazolam and vancomycin [10],suggesting a low risk of incompatibility with rhIGF-1/rhIGFBP-3;however,particular attention was given to the small-molecule concentrations that were assessed in regard to compatibility with insulin.No compatibility data are available to date for insulin with fentanyl or fluconazole;however,on the basis of theoretical evaluation and the solution pH,a reaction is not expected.
Among the drugs that were observed to be incompatible with rhIGF-1/rhIGFBP-3 in the in vitro testing studies,the risk was classified as medium/high and appropriate actions were recommended.An example of a risk assessment,for morphine,is provided in Supplementary Table 2 in the Supplementary Information.

Table 4 Physical and small-molecule compatibility of the rhIGF-1/rhIGFBP-3 drug product and small-molecule test medications

Table 4 (continued)
Discussion
In this study,in vitro testing indicated the physical compatibility of the rhIGF-1/rhIGFBP-3 drug product with 11/19 medications and nutritional therapies under the conditions and doses tested.For medications showing in vitro compatibility,the risk evaluation confirmed a low probability and severity of risk for incompatibility.Physical compatibility was not established with 8/19 medications.For drugs identified as incompatible,infusions would need to be redistributed to optimize available IV lines.
Historically,there has been a lack of compatibility data available for drugs administered to preterm infants.A review of neonatal drug studies found that no documentation on compatibility was available for almost 60% of IV drug-drug infusions,and for 34% of IV drug-nutrition coinfusions administered in the NICU [20].
As might be expected,there is a lack of comprehensive drug testing in neonates.One US study reported that only 35% of medications administered to neonates were approved by the US Food and Drug Administration for use in infants[21],and an Italian study found that 44% of medications were prescribed off-label in the preterm neonatal population [22].Such findings have potential implications for both treatment efficacy and safety in infants.The majority of compatibility studies are performed for small molecules coadministered with small molecules;literature was identified for compatibility testing for only two biologics,insulin and vasopressin [12,14],and standard biologic-specific testing methods have not been established.
In this study we investigated the physical compatibility of the rhIGF-1/rhIGFBP-3 drug product when mixed with frequently administered medications.To generate a complete picture of compatibility,we evaluated chemical compatibility at the level of the small-molecule content as part of a separate study on the protein content/chemical modification[18].It should be noted that the main challenge with the protein-specific methodologies is the extremely low protein concentration of rhIGF-1/rhIGFBP-3 post mixing with each small molecule.The low concentrations require highly specific and robust analytical methods.
rhIGF-1/rhIGFBP-3 is being investigated for the prevention of complications of prematurity,with an ongoing comprehensive clinical trials program to evaluate the safety and efficacy of the drug.The present work is being conducted as part of this program,to systematically evaluate and build a comprehensive body of data on the compatibility of rhIGF-1/rhIGFBP-3 with commonly administered intravenous drugs.Our goal is eventually to evaluate a large panel of medications and to build up a comprehensive body of data to aid clinicians’ decision-making regarding the coinfusion of rhIGF-1/rhIGFBP-3.Drug compatibility testing studies should be conducted as early as feasible in the investigational phase of a neonatal drug to allow sufficient time for study findings to inform clinical trials as well as the eventual adoption of the drug in clinical practice.
An inherent limitation of the current study is the fact that the results are based on in vitro mixing under specific controlled conditions,and the risk evaluation is theoretical.Careful monitoring will be required in the clinical setting.The risk assessment will evolve when more protein-specific data become available.For example,recent protein-specific analyses demonstrated that the presence of the excipient sodium (meta)bisulfite is damaging to rhIGF-1/rhIGFBP-3 owing to its oxidation.Therefore,although no physical (pH) change was observed for the dobutamine and norepinephrine mixtures,these medications have been removed from the compatible list because they are formulated with sodium (meta)bisulfite.Additionally,the presence of (meta)bisulfite will be added to the risk analysis strategy.
In conclusion,the administration of rhIGF-1/rhIGFBP-3 with various medications will be facilitated by conducting selected in vitro studies and case-by-case risk assessments.This study adds a novel approach to the compatibility testing of drugs utilized in the NICU and may set a benchmark for future studies.Early contributions from clinicians and cross-functional disciplines will be essential to the process.
Supplementary InformationThe online version contains supplementary material available at https:// doi.org/ 10.1007/ s12519-022-00610-9.
AcknowledgementsParts of these data have been presented at the joint European Neonatal Societies 1st Congress (September 16–20,2015,Budapest,Hungary),and at Hot Topics in Neonatology 2015(December 6–9,2015,Washington,DC,USA).The authors thank the following people for their contributions to the development of this study: Sujit Basu,PhD,Helge Reisch,PhD,and Katherine Taylor,PhD,employees of Takeda Pharmaceutical Company Limited;Bing He,PhD,and Nerissa Kreher,MD,MBA,employees of Takeda Pharmaceutical Company Limited at the time of the study design;and the analysts at PPD (specifically Mark Ross) and staffat Tufts University.The authors also thank Tracy Hsaio,employee of Takeda Pharmaceutical Company Limited at the time of the study,for her significant role in the development of (Fig. 1) and the overall risk assessment strategy.Under direction of the authors,Rosalind Bonomally,MSc,employee of Excel Medical Affairs,provided writing assistance for this manuscript.Editorial assistance in formatting,proof reading,and copy editing also was provided by Excel Medical Affairs.Takeda Pharmaceutical Company Limited provided funding to Excel Medical Affairs for support in editing this manuscript.The interpretation of the data was made by the authors independently.
Author contributionsAll authors contributed to the interpretation of data from their perspective of subject matter experts in various relevant fields of biologic and small-molecule formulation and stability,clinical operations,and clinical neonatology.All authors reviewed and edited each draft of the manuscript and read and approved the final draft.
FundingThis study was funded by Takeda Pharmaceutical Company Limited.
Data availabilityThe datasets,including the redacted study protocol,supporting the results reported in this article will be made available within three months from initial request to researchers who provide a methodologically sound proposal.
Declarations
Conflicts of interestNS-M,ND,and ST are employees and stockholders of Takeda Pharmaceutical Company Limited.AB,EJ,SS,YSH and NB were employees of Takeda Pharmaceutical Company Limited at the time of the study.MAT’s employing institution has a consultancy agreement with Takeda relating to his involvement with this product.BM is the CEO of Paidion Research.CM has no competing interests to disclose.
Ethical approvalNot applicable for this study not involving humans or animals.
Open AccessThis article is licensed under a Creative Commons Attribution 4.0 International License,which permits use,sharing,adaptation,distribution and reproduction in any medium or format,as long as you give appropriate credit to the original author(s) and the source,provide a link to the Creative Commons licence,and indicate if changes were made.The images or other third party material in this article are included in the article's Creative Commons licence,unless indicated otherwise in a credit line to the material.If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use,you will need to obtain permission directly from the copyright holder.To view a copy of this licence,visit http:// creat iveco mmons.org/ licen ses/ by/4.0/.
 World Journal of Pediatrics2023年1期
World Journal of Pediatrics2023年1期
- World Journal of Pediatrics的其它文章
- Innovative treatments for congenital heart defects
- Perioperative extracorporeal membrane oxygenation in pediatric congenital heart disease: Chinese expert consensus
- Role of ultrasound in the treatment of pediatric infectious diseases:case series and narrative review
- Manual and alternative therapies as non-pharmacological interventions for pain and stress control in newborns: a systematic review
- Vitamin D therapy in pediatric patients with inflammatory bowel disease: a systematic review and meta-analysis
- Development of necrotizing enterocolitis after blood transfusion in very premature neonates