
线粒体ALDH2通过调控自噬对缺氧性肺动脉高压的保护机制研究
2023-02-24 01:27李小荣陈永锋康品方王洪巨
蚌埠医学院学报 2023年1期
李小荣,鲜 维,2,谭 鑫,陈永锋,唐 碧,张 恒,康品方,王洪巨
肺动脉高压(PAH)是一种慢性进行性疾病,是一种被定义为在海平面,静息状态下通过右心导管测定的平均肺动脉压超过25 mmHg的疾病,世界上大约有1%的人口都受到了PAH的影响[1-2]。缺氧性肺动脉高压(hypoxic pulmonary hypertension,HPH)是其中第三类,它主要是由慢性缺氧所导致的例如慢性阻塞性肺疾病等。HPH的主要特征表现为肺血管的阻力增加,最终导致右心衰竭甚至死亡,是一种预后极差、对医疗负担极重的疾病[3-5]。远端小动脉是HPH的主要靶点,它的主要病理特征是肺血管的重构,血管壁表现出促增殖/抗凋亡和高代谢状态,毛细血管前小动脉肌化,并随着时间的推移导致血管闭塞。其中血管内皮损伤及平滑肌细胞增殖在肺血管的重构中发挥着中重要作用[6-7]。目前虽然已经开发了多种治疗PAH的药物,但重度肺动脉高压病人的长期预后仍然很差[8-9]。因此,我们亟需更多的研究靶点以求改善HPH预后并减轻医疗负担。
线粒体乙醛脱氢酶(ALDH2)隶属于乙醛脱氢酶家族,是一种位于线粒体基质中的变构四聚体酶,最早在肝脏中被发现,后续研究[10-12]发现在心脏、大脑、肠道等多种器官中也大量表达。在多项研究中发现ALDH2通过清除脂质过氧化产生的有毒醛,特别是4-HNE,在心肌损伤和中风中发挥着保护作用。有研究证实ALDH2激活可减轻野百合碱诱导的PAH[13-14],同时,XU等[15]研究发现ALDH2可能通过调控4-HNE的水平减轻HPH的发生。
自噬是将细胞质物质传递到动物细胞中的溶酶体或植物和酵母细胞中的液泡的所有途径的总称。主要可分为三类:巨自噬、微自噬以及分子伴侣介导的自噬。自噬分为三个过程:自噬起始、自噬膜延伸和自噬溶酶体形成[16-18]。Beclin1是自噬过程中的关键蛋白之一,在自噬过程中,Beclin1在调节自噬小体膜的形成和物质转运过程中起着重要作用。Beclin1主要通过Atg14L-Vps15-Vps34信号轴调控巨自噬的起始[19]。微管相关蛋白 1A/1B 轻链 3B(MAP1 LC3)是微管相关蛋白,它们介导微管和细胞骨架成分之间的物理相互作用,通过泛素样修饰参与自噬体液泡(自噬体)的形成。并且对于自噬体成熟的后期阶段至关重要,同时它将线粒体消除降低到基础水平以满足细胞能量需求并防止过量活性氧自由基(ROS)产生从而参与线粒体自噬以调节线粒体的融合与分裂[20-21]。SQSTM1(p62)是巨自噬所需的自噬受体。作为多泛素化自噬小体和溶酶体之间的桥梁。直接与降解的物质和 MAP1 LC3 家族的自噬修饰剂相互作用,在自噬溶酶体的形成中发挥了关键作用[22-23]。最近有研究表明,缺氧能够通过促进自噬的发生进而激动肺动脉平滑肌细胞(PASMC)的增生与迁移,并且抑制自噬能够有效的减缓野百合碱诱导的PAH的发生发展。
本研究拟用通过慢性缺氧构建HPH动物模型,同时使用Alda-1以及Daidzin分别激动、抑制线粒体ALDH2,观察肺组织病理改变,同时建立缺氧性肺动脉高压细胞模型,观察PASMC细胞中自噬水平的变化,同时用CCK8检测细胞的增殖水平、流式细胞术测定细胞凋亡,从而探究ALDH2、自噬与HPH的关系,为临床通过ALDH2抑制自噬从而改善甚至治疗HPH提供新的方法及思路。
1 材料和方法
1.1 动物和试剂 SD大鼠由蚌埠医学院提供且符合动物伦理;胰酶消化液(Gibco);特级胎牛血清(BI);DMEM高糖培养基(Gibco);双抗(Biosharp);PBS缓冲液(Biosharp);Alda-1(SML0462,Sigma-Aldrich);Daidzin(Sigma-Aldrich);ALDH2、Beclin、BLC3β、p62抗体购自ABCAM;β-actin购自Affinity;BCA试剂盒(碧云天);T25培养瓶(Corning)。
1.2 方法
1.2.1 大鼠HPH模型构建 将雄性SPF级SD大鼠随机分为4组:常氧组(N组)、常氧+Alda-1灌胃组(N+Alda-1组)、缺氧组(H组)、缺氧+Alda-1灌胃组(H+Alda-1组),每组6只,按照昼12 h夜12 h规律光照培养,缺氧组以8 h/d,5天/周的10%±0.5%氧浓度进行缺氧处理,4周过后游离出左下肺进行HE染色观察病理变化。
1.2.2 形态学检测 将右下肺用PBS冲洗干净后,4%多聚甲醛固定,然后依次使用低-高浓度乙醇脱水,再用二甲苯脱蜡,石蜡包埋。使用切片机(徕卡)切成约4 μm厚度切片,进行HE染色,使用显微镜观察并记录,扫描并拍照观察肺血管形态。
1.2.3 大鼠肺动脉平滑肌细胞原代培养 取出生4~8周的SPF级SD大鼠,颈椎脱位法处死后用75%乙醇浸泡消毒10 min,然后将大鼠固定于紫外灭菌好的超净工作台中,用高压灭菌的器械迅速分离出左下肺,用无菌PBS冲洗净血液后固定在装有无菌PBS的6 cm培养皿中。在体视显微镜下用无菌的眼科剪及眼科镊小心分离出三四级肺动脉,去除外膜后置于新鲜PBS中,用眼科剪剪开血管,用无菌棉签或者无菌手术刀轻轻刮去内膜,并用眼科剪将血管在血清中剪成1 mm3大小。将血管组织块均匀的分布到T25培养瓶中,缓慢吸出血清,将培养瓶倒置2 h后加入20%的完全培养基,约48 h可见组织周围有梭形平滑肌爬出。
1.2.4 肺动脉平滑肌细胞鉴定 用免疫荧光对培养的肺动脉平滑肌进行鉴定。将培养瓶中的PASMCs消化后,取4万细胞接种于48孔板大小的爬片中,次日可进行免疫荧光实验。用无菌PBS清洗细胞2次,1分钟/次;4%的多聚甲醛室温固定10 min,再用PBS洗3次,5分钟/次;然后用5%BSA配制的0.3%的TritonX-100对细胞进行通透及封闭1 h;用上述封闭液稀释的α-横纹肌肌动蛋白一抗过夜孵育PASMCs;次日用PBS清洗细胞3次,5分钟/次;用封闭液配制的羊抗小鼠的FITC标记的二抗避光孵育1 h;在避光条件下用PBS清洗细胞3次,5分钟/次;将细胞爬片取出用滤纸吸取残余液体,细胞面朝下盖在滴加了含DAPI的抗荧光淬灭剂的载玻片上,封片后避光下用荧光显微镜观察。
1.2.5 细胞实验分组 细胞实验分为6组:常氧组(N组)、常氧+Alda-1组(NA组)、缺氧组(H组)、缺氧+Alda-1组(HA组)、缺氧+Daidzin组(HD组)、缺氧+Alda-1+Daidzin组(HAD组)。
1.2.6 CCK8 取3 000对数生长期的PASMCs接种于96孔板中,干预后的细胞培养基中,按照体积100∶10的比例加入CCK8试剂,37 ℃孵育1 h后,利用酶标仪于450 nm处测定吸光度,使吸光度值保持在1.0~2.0。
1.2.7 Annexin V/PI测定细胞凋亡 取10万对数生长期的PASMCs接种于6孔板中,额外种3孔以做对照,干预结束后,使用1 mL 0.25%胰酶消化细胞,加入2 mL完全培养基终止消化后,轻轻吹打使细胞脱落,转移到15 mL离心管中,4 ℃、1 000 r/min离心5 min,去上清加入预先配置的Annexin V/PI室温孵育30 min后,放置于冰上,使用流式细胞仪(FACSCalibur,美国BD公司)测定细胞凋亡水平。
1.2.8 Western blotting检测ALDH2、p62、LC3β蛋白表达情况 干预后的细胞用配置了1 mmol/L的PMSF的PBS洗涤后,向T25培养瓶加入1 mL 0.25%胰酶消化1 min,用3 mL 10%的完全培养基终止消化,将混合液离心,4 ℃ 1 000 r/min离心5 min;在沉淀中加入适量含有1 mmol/L PMSF的RIPA裂解液,冰上裂解30 min后离心,4 ℃ 12 000 g离心15 min;取上清,根据BCA蛋白定量试剂盒说明书测定蛋白浓度;制备10%的聚丙烯酰胺凝胶,每孔上样30 μg;浓缩胶用80 V恒压电泳30 min,分离胶用120 V恒压电泳90 min;将电泳好的凝胶置于三明治夹板中转膜,220 mA恒流90 min电转到PVDF膜上;5%脱脂奶粉室温封闭90 min;用5%脱脂奶粉稀释的一抗于4 ℃摇床孵育过夜;次日用TBST清洗条带4次,5分钟/次;用5%脱脂奶粉稀释的二抗室温孵育2 h,TBST清洗3次,10 分钟/次;用ECL试剂盒曝光显影。
1.3 统计学方法 采用单因素方差分析(ANOVA)对多组间均数进行比较。
2 结果
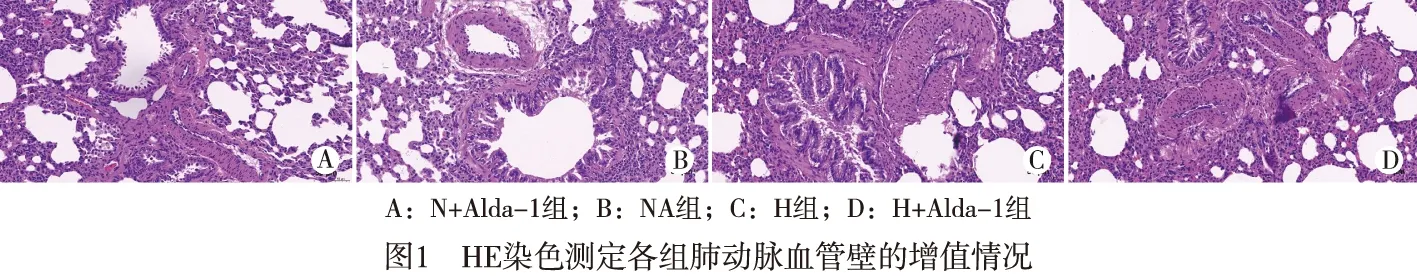
2.1 肺组织病理改变 HE染色结果发现,通过慢性缺氧处理过后,HC组的肺动脉血管壁明显增厚,可见中膜增厚,管腔变窄;通过Alda-1干预处理后,H+Alda-1组的血管壁厚度明显减轻,同时缺氧所致的管腔狭窄也有所恢复(见图1)。
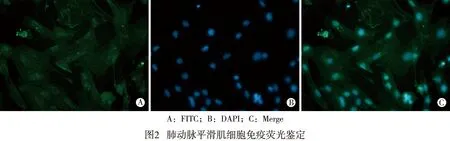
2.2 原代肺动脉平滑肌细胞免疫荧光鉴定结果 PASMC免疫荧光显示细胞核被DAPI染为蓝色,α-SMA着色为绿色(见图2)。结果可见,α-SMA阳性细胞率较高,提示细胞纯度可用于后续细胞实验。
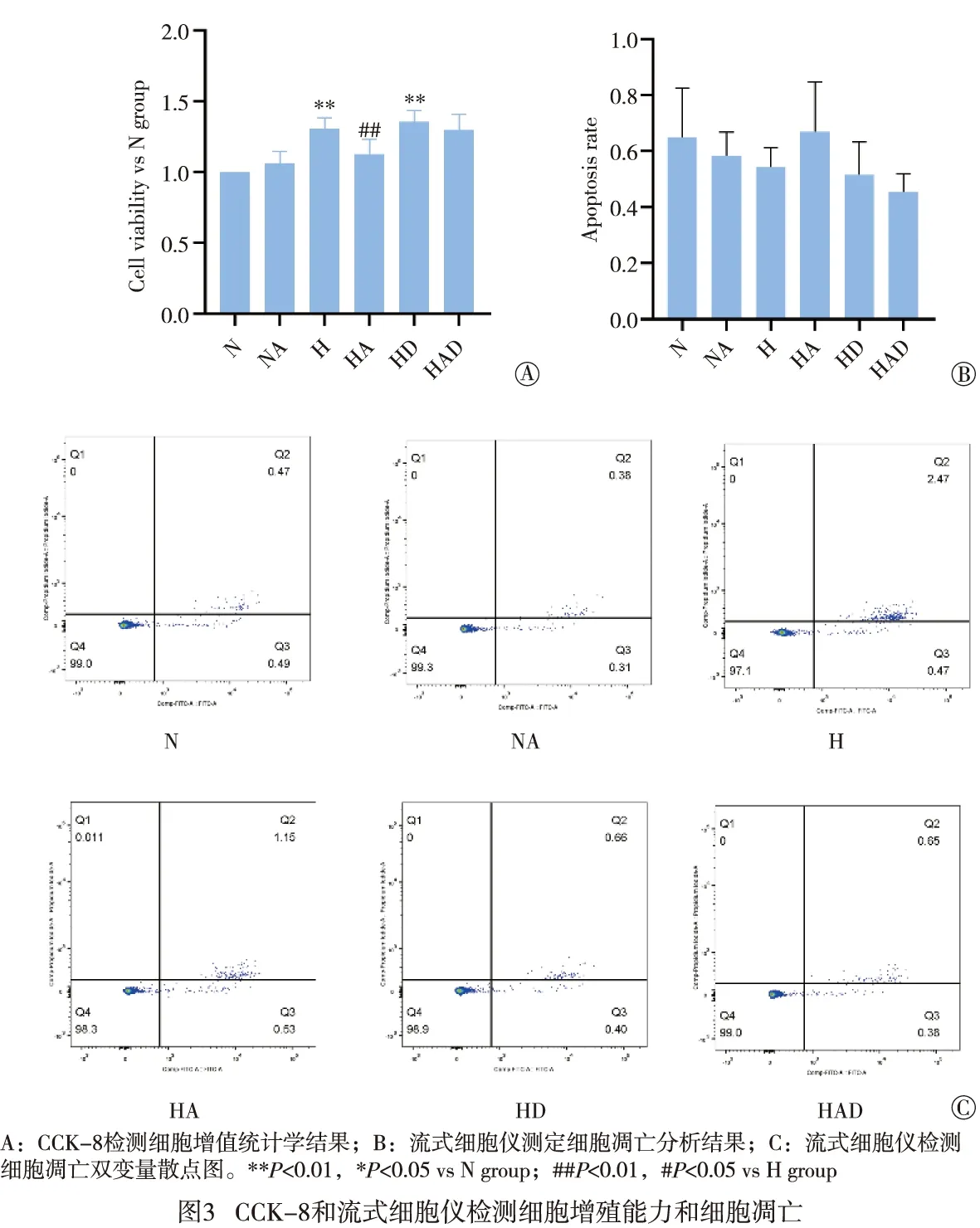
2.3 CCK8结果 CCK-8结果显示,与N组相比,H组与HD组的细胞活性与增殖明显增强(P<0.01);与H组相比,HA组细胞增殖有所减少(P<0.01),HAD组细胞增殖无统计学意义(P>0.05)(见图3A)。
2.4 细胞凋亡水平 流式细胞术检测结果发现,与N组相比,H组细胞凋亡有少许增加,但差异均无统计学意义(P>0.05);同时与H组相比,HA组和HAD组细胞增殖有所减少,但差异均无统计学意义(P>0.05)(见图3B、C)。
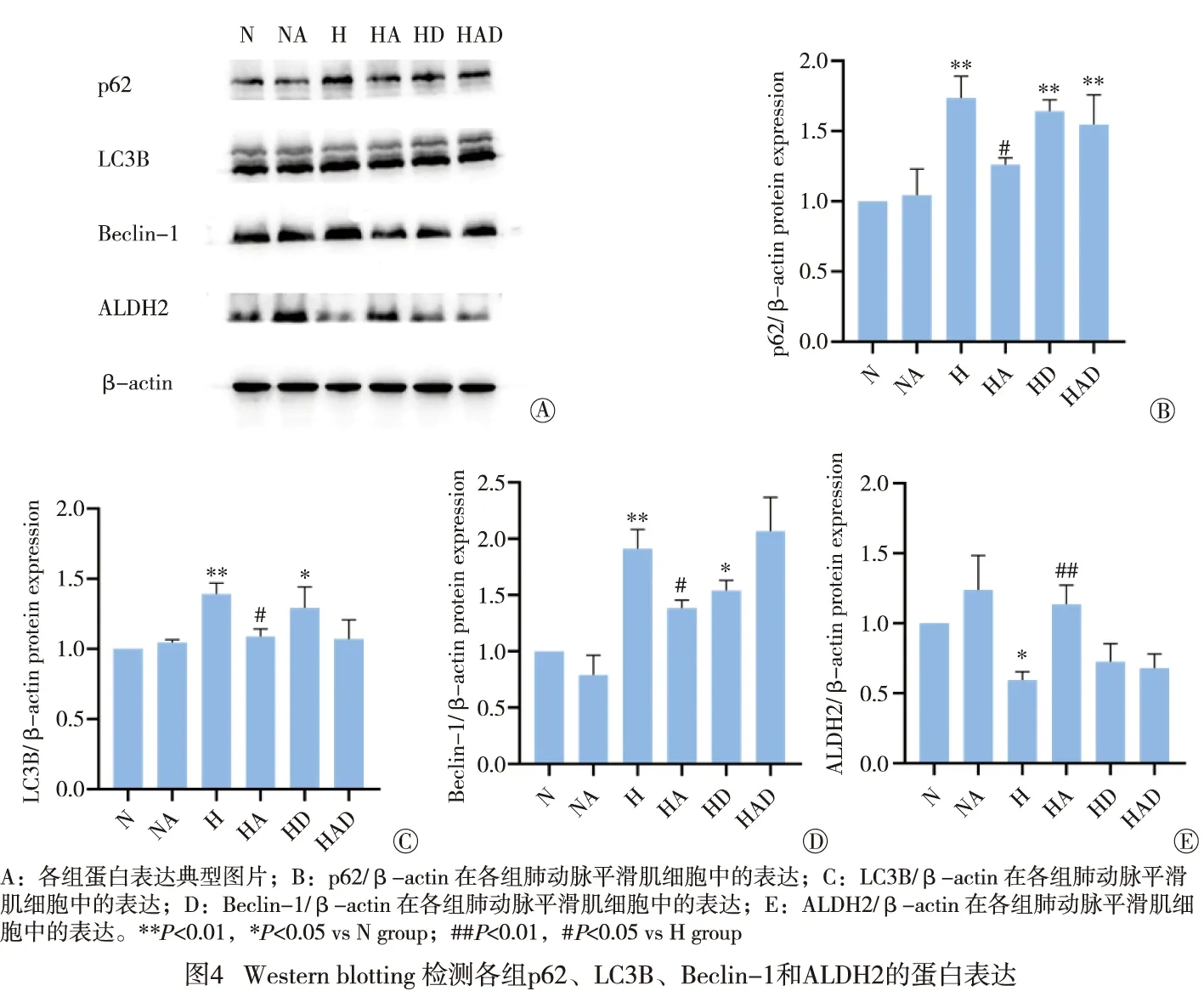
2.5 Western blotting检测ALDH2、p62、Beclin1、LC3B蛋白表达情况 通过Image J对蛋白条带进行分析,与N组相比,H组与HD组的ALDH2表达降低(P<0.05),自噬相关蛋白p62、Beclin1、LC3B均明显升高(P<0.01);与H组相比,HA组ALDH2表达明显升高(P<0.01),自噬相关蛋白p62、Beclin1、LC3B均降低(P<0.05),HAD组的Beclin、LC3B蛋白差异无统计学意义(P>0.05)(见图4)。
3 结论
肺小动脉缺氧是PAH形成的重要因素,但其机制尚不完全明确[6]。在近年的研究中发现,PAH的主要病理变化体现在肺动脉中内皮细胞功能障碍与肺动脉平滑肌的异常增殖与收缩。其中在HPH中,肺动脉中膜中PASMCs的增殖是肺血管重构的最主要因素[24]。ALDH2是一种非细胞色素P450线粒体醛氧化酶,许多心血管疾病与氧化应激和线粒体功能障碍有关。在ZHAO等[25]研究中发现,在慢性缺氧中,敲除ALDH2会加重小鼠的HPH进展,并且ALDH2可能通过调控线粒体的分裂与融合对PASMCs的增殖有着一定的调控作用。因此,本研究拟通过慢性缺氧构建大鼠HPH动物模型,缺氧诱导原代培养的SD大鼠肺动脉平滑肌细胞构建HPH的细胞模型,以探讨在HPH中PASMC增殖于凋亡失衡的可能因素。本实验结果发现,在动物模型中,与N组相比,通过慢性缺氧处理后,肺组织中肺小动脉血管管壁厚度明显增加,尤以中膜为著,血管管壁也因此变窄;而与H组相比在Alda-1激活线粒体ALDH2后,病理切片结果显示肺小动脉血管管壁厚度明显出现了明显降低,并且因缺氧所导致的官腔狭窄也有所减轻。这些实验结果提示慢性缺氧处理能够在大鼠中复制出HPH的模型,同时通过Alda-1激活线粒体ALDH2能够对HPH发挥一定的治疗效果。
有研究发现在MCT诱导的PAH中,AMPK 介导的mTOR 自噬信号通路参与细胞自噬的调节。在缺氧性肺动脉高压中,自噬水平也明显升高[26-27]。这些结论与我们的实验结果相符,经过缺氧处理后,PASMCs中的ALDH2水平明显降低,自噬相关蛋白p62、Beclin1及MAP1 LC3B水平显著升高,说明自噬在PAH的发病过程中发挥了重要作用。同时在Alda-1组孵育过后,ALDH2水平升高,自噬相关蛋白p62、Beclin1及MAP1 LC3B的表达均有下降,提示激活ALDH2能够有效降低PAH中自噬的水平。在既往自噬的研究中,往往认为Beclin1及LC3B参与了自噬的发生与自噬膜的延长,并且该蛋白的表达水平与自噬水平呈现正相关关系,而p62参与自噬中自噬小体与溶酶体的结合以及降解的过程,该蛋白的表达量与自噬水平呈现负相关关系,本研究中的结果显示,在缺氧时PASMCs中Beclin1、LC3B及p62水平均升高,提示可能缺氧可以促进PASMCs的自噬发生,但是对自噬溶酶体的降解产生了抑制。
研究表明,适度的自噬可以导致细胞迁移和平滑肌细胞的增殖,用声波刺猬蛋白治疗平滑肌细胞可诱导自噬并增加细胞增殖;此外,通过PDGF诱导的自噬与平滑肌细胞的增殖与迁移密切相关,同时,利用3-MA抑制自噬可以减少细胞的增殖[28-29]。在本研究中发现,与常氧组相比,缺氧处理后的细胞内自噬水平明显升高,同时CCK8与流式细胞术结果显示,通过缺氧处理后,PASMCs的活性与增殖能力明显增加,凋亡水平虽有所降低,但变化并无统计学意义;而与缺氧组相比,在通过Alda-1激活线粒体ALDH2后,细胞内自噬水平显著降低,PASMCs的增殖也明显受到抑制,细胞凋亡虽有所增加,但是通过统计分析,差异并无统计学意义。这些结果提示线粒体ALDH2可能通过对自噬水平的调控,进而主要通过降低PASMCs的增殖水平而非调控凋亡的水平,从而改善HPH的病理改变。
综上所述,线粒体ALDH2可能通过对自噬的调控,进一步调节了PASMCs的增殖,进而改善HPH的病理进程,而通过激活线粒体ALDH2在一定程度上可以改善肺动脉高压的发生发展。
猜你喜欢
昆明医科大学学报(2021年6期)2021-07-31
心肺血管病杂志(2020年5期)2021-01-14
中国临床医学影像杂志(2019年4期)2019-06-18
浙江中西医结合杂志(2018年12期)2018-12-27
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
罕少疾病杂志(2016年5期)2016-03-11
中国体外循环杂志(2015年3期)2015-12-08
华南农业大学学报(2015年5期)2015-12-04
郑州大学学报(医学版)(2015年1期)2015-02-27
