
内蒙古赛罕乌拉自然保护区马鹿夏季肠道菌群多样性
2023-02-22 02:49刘夫仁张正一冯中华张梓琪鲍伟东
野生动物学报 2023年1期
刘夫仁,赛 罕,贺 伟,张正一,冯中华,张梓琪,鲍伟东
(1.北京林业大学生物科学与技术学院,北京,100083;2.山东省枣庄市山亭区农业农村局,山亭,277200;3.内蒙古赛罕乌拉国家级自然保护区管理局,大板,025150;4.赤峰市森林草原保护发展中心,赤峰,024005;5.赤峰市野生动植物保护协会,赤峰,024005)
动物体内肠道中有数以万亿计的菌群,不同季节、饮食和栖息地的变化都会导致肠道菌群组成发生变化,达到动态平衡[1-3]。肠道菌群与宿主存在相互影响,在不同物种之间肠道菌群也会发挥不同作用,如食草动物粪便中分离出的菌株多具有纤维素分解功能,而从食肉动物粪样中分离出的菌株多具有分解脂肪的功能,同时肠道菌群也会反作用于宿主本身,促进物种对栖息地、食性的选择[4]。Li 等[5]的研究发现肠道菌群还可以扩大动物的食物生态位,某些植物为防止被动物取食,会合成一些次生化合物,而动物肠道菌群则可以消化这些化合物。
食性是影响野生动物肠道菌群的重要因素,不同的食物组成会影响宿主肠道菌群的多样性及其群落结构,可以反映物种对环境的适应程度。在雌雄性坡鹿(Rucervus eldii)的研究中发现,不同性别之间肠道菌群组成无明显差异,但菌群丰度存在差异,这种结果主要是由食物不同造成的[6]。对野生和圈养林麝(Moschus berezovskii)肠道菌群的研究发现,野生个体和圈养个体肠道群落存在显著差别,圈养林麝拥有更高丰度的变形菌门(Proteobacteria)[7]。
马鹿(Cervus elaphus)生活于范围较大的针阔混交林、林缘或靠近水源的林地,是国家二级重点保护野生动物[8]。对于马鹿的生态研究涵盖了食性、家域、种群遗传结构和行为习性等多方面内容[9-12]。阿拉善马鹿(C.e.alashanicus)冬季肠道菌群以厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)和变形菌门为优势菌门,显示菌群群落组成与食性密切相关[13]。不同性别东北马鹿(C.e.xanthopygus)的冬季菌群多样性无显著差异[14]。在内蒙古赛罕乌拉国家级自然保护区有蹄类动物中,马鹿的种群数量较多[15],其他有蹄类动物还包括野猪(Sus scrofa)、狍(Capreolus pygargus)和中华斑羚(Naemorhedus griseus)等[16]。近年来,随着保护政策的落实,野生动物保护成效得到极大提升,各类动物种群密度增加,这导致不同物种在生态位中存在激烈竞争,了解不同物种的适应对策,有助于改进保护管理策略。本研究旨在分析保护区内夏季马鹿的肠道菌群多样性及结构,从食物资源利用和动物健康角度认识马鹿的适应性,为后期开展同域分布的有蹄类物种资源利用及竞争研究提供基础数据,同时也为马鹿的保护提供新视角。
1 研究方法
1.1 样品采集
样品采集于内蒙古赛罕乌拉国家级自然保护区,该保护区位于内蒙古自治区赤峰市巴林右旗北部(43°59'—44°27' N,118°18'—118°55' E),属于大兴安岭南段山地,占地总面积10.04万 hm2[17]。2020年7—8 月,在保护区正沟、大东沟和大西沟按照样线法取马鹿粪便样品35份(图1),样线间距500 m左右,每条样线只取1份样品(10粒),记录粪便样品形状、大小和位置信息,样品放入收集管中倒入95%乙醇冷冻保存。
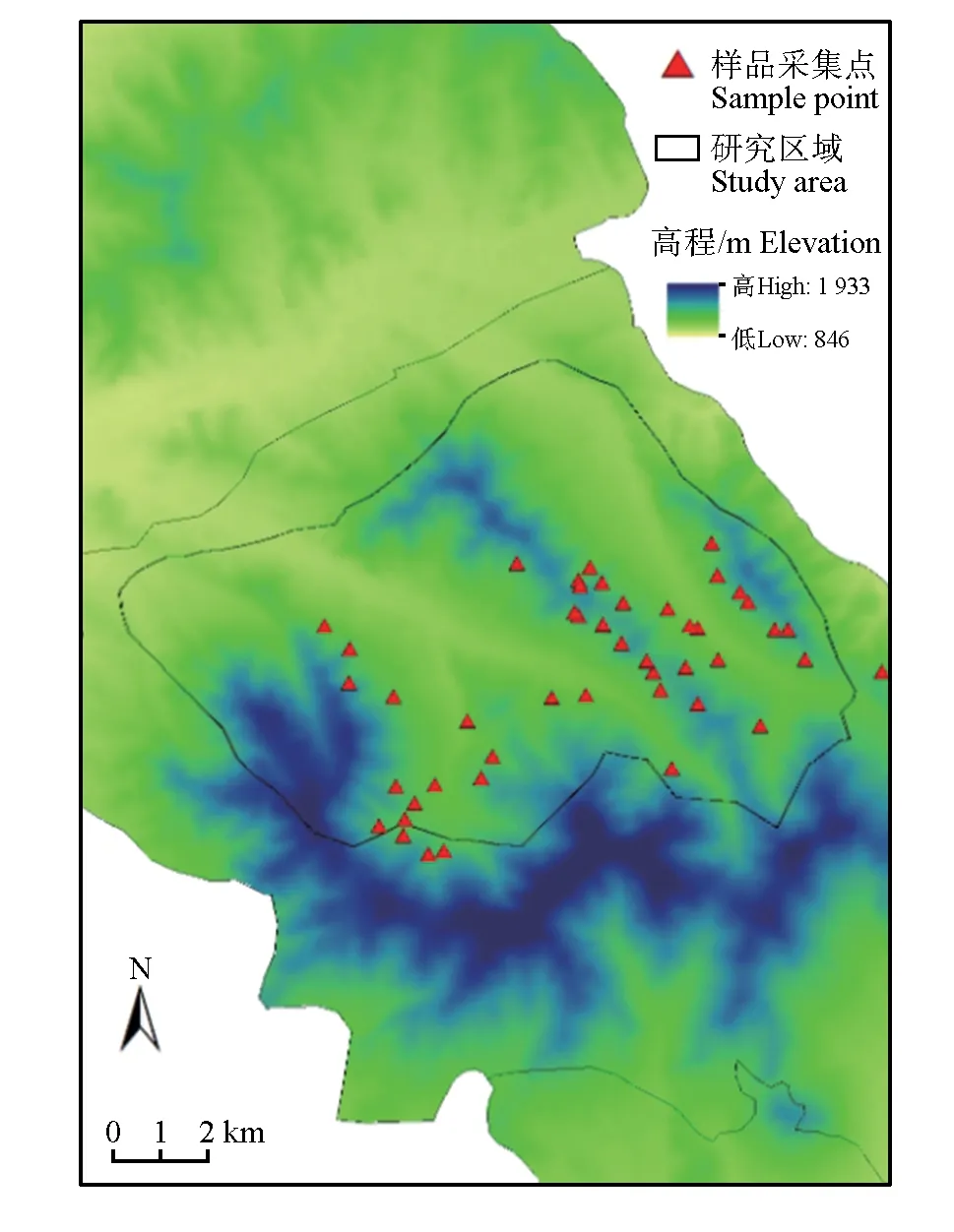
图1 马鹿肠道菌群多样性分析粪样采集点Fig.1 Pellets sampling sites for red deer gut microbiota diversity analysis
1.2 DNA提取及测序
使用E.Z.N.A 粪便DNA 提取试剂盒(Omega Bio-Tek)从粪便样本中提取总DNA。使用肠道细菌通用引 物343F:5'-TACGGRAGGCAGCAG-3' 和798R:5'-AGGGTATCTAATCCT-3' 扩增细 菌16S rRNA 基因V3—V4 高变区。通过PCR 反应向16S rRNA 的产物末端加上带有Index 的接头,用于Illumina MiSeq 的NGS 测序。PCR 反应体系为25.0 µL:模板DNA 20.0 ng,dNTPs 2.0 µL,引物各1.0 µL(10 µmol/L),TransStart Buffer 2.5 µL,TransStartTaqDNA 0.5 µL,用ddH2O 补足。PCR 反应设置:94 ℃预变性3 min;94 ℃变性5 s,57 ℃退火90 s,72 ℃延伸10 s,24 个循环;72 ℃终延伸5 min。通过Qubit 3.0荧光计检测DNA 样品的浓度,采用MetaVx文库试剂盒构建DNA文库。
1.3 数据分析
使用QIIME 数据包分析16S rRNA 测序数据,使用UCHIME 去除嵌合序列,参考数据库Silva 128 比对16S rRNA 序列,使用聚类程序Vsearch(1.9.6)将序列聚类为操作分类单位(OTU),序列相似性阈值设为97%。基于OTU 分析的结果,采用对样本序列进行随机抽样的方法,以QIIME软件计算Alpha多样性指数(Chao1、Shannon和Simpson指数)以反映肠道微生物群落的物种丰度和多样性,采取Good’s coverage 曲线用于评估测序结果的群落物种总数,判断测序深度是否足够。使用unweighted UniFrac 比较分析样本间是否有显著差异,基于Bray-Curtis 样本间距离矩阵用于主坐标分析(PCoA)可视化图展示Beta多样性,并进行非度量多维尺度(NMDS)排序。
2 结果
2.1 测序结果
经过对16S rRNA 基因测序数据分析,35份马鹿样品共获得2 287 019 条有效序列,其中最大有效序列数为70 650,最小有效序列数为50 488,每个样品平均获得有效序列为65 343。对有效序列进行聚类分析,OTU 平均数量为2 553,其中核心OTU 数量为169(图2)。Good’s coverage 曲线显示测序深度平均达到99%以上,说明测序深度足以充分反映样本真实性,可以进一步分析(图3)。
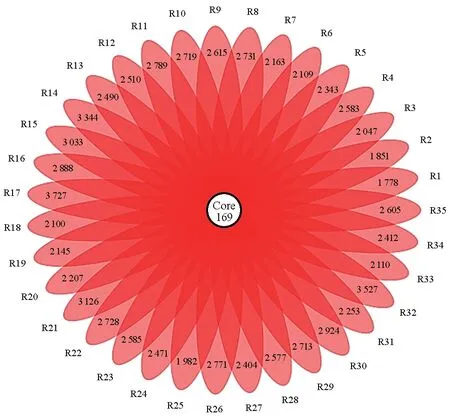
图2 马鹿粪便样品OTU的Venn图Fig.2 Venn diagram of red deer fecal sample OTU
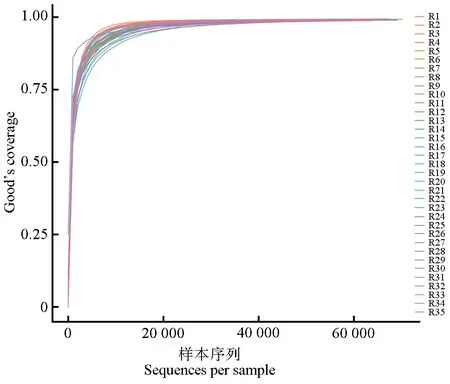
图3 马鹿粪便样品Good’s coverage曲线Fig.3 Good’s coverage curve of red deer fecal samples
2.2 多样性分析
Chao1指数在样本序列达到20 000时,曲线趋于平缓,Chao1 最高值为4 358,最低值为2 648(图4A);Shannon 指数在样本序列为4 000 时,曲线趋于平缓,最高值为9.9,最低值为6.3(图4B);Simpson指数在样本序列达到2 300 时,曲线趋于平缓,最高值为0.99,最低值为0.96(图4C)。结果表明,样品的丰富度和多样性都较高。
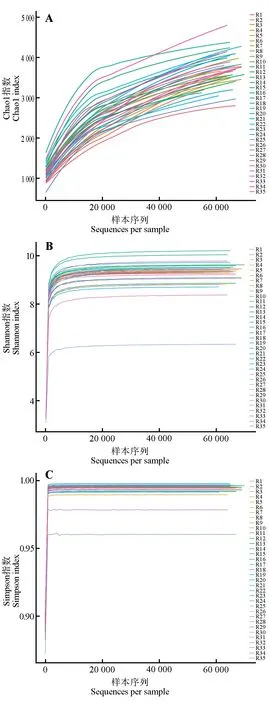
图4 马鹿肠道菌群Alpha多样性Fig.4 Alpha diversity of red deer gut bacteria community
NMDS stress 数值小于0.2,除一份样品(R25)可能存在测序深度不足外,其余样本分布较均匀,个体无明显差异(图5A)。在PCoA 分析中,样本分布较均匀,除R25 有明显分隔,其余个体无明显分离,种内并无显著差异(图5B)。
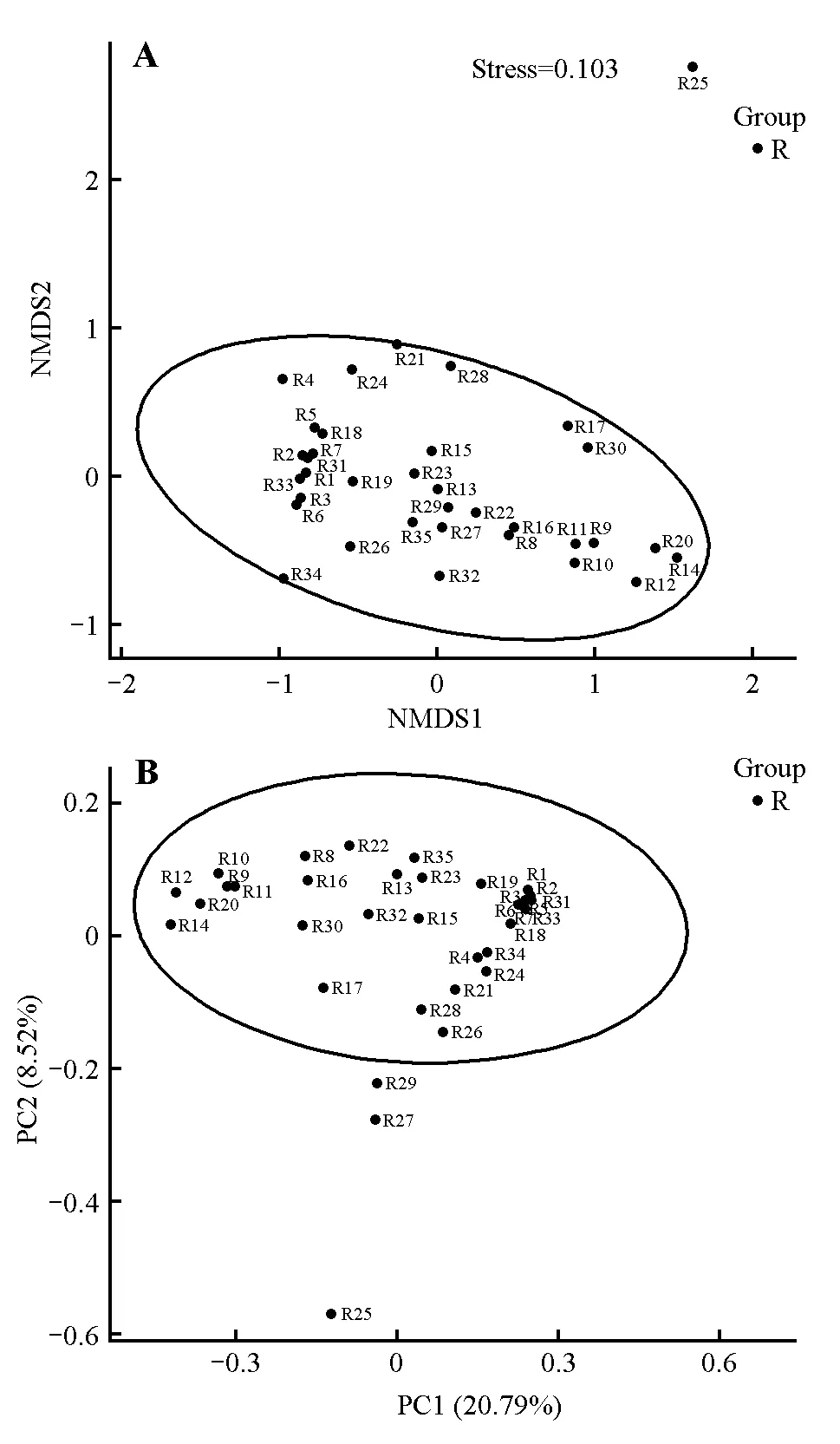
图5 马鹿肠道菌群β多样性Fig.5 Beta diversity of red deer gut bacteria community
2.3 群落结构分析
通过对OTU 聚类注释分析,35 份马鹿粪便样品中共获得38 菌门、777 菌属,用相对丰度描述肠道菌群的组成。在门水平,相对丰度之和>95%的是拟杆菌门(29.55%)、变形菌门(29.30%)、厚壁菌门(29.30%)、放线菌门(Actinobacteria,5.78%)和芽单孢菌门(Gemmatimonadetes,1.99%),其余门水平的肠道菌群相对丰度较低(图6)。
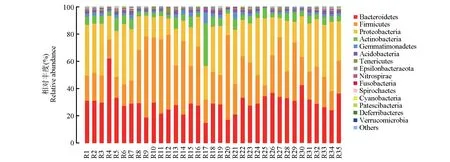
图6 马鹿肠道菌群门水平相对丰度Fig.6 Relative abundance of gut bacteria composition at the phylum level
在属水平,取相对丰度前15的菌属进行作图,其中丰度超过1%的菌属共11种,分别是拟杆菌属(Bacteroides,4.30%)、瘤胃球菌属(Ruminococcaceae_UCG-005,3.34%)、埃希氏菌-志贺氏菌属(Escherichia-Shigella,2.87%)、柠檬酸杆菌属(Citrobacter,2.62%)、毛螺菌属(Lachnospiraceae_NK4A136_group,2.43%)、瘤胃球菌属(Ruminococcaceae_UCG-014,1.98%)、另枝菌属(Alistipes,1.88%)、普雷沃菌属(Prevotella_1,1.70%)、乳杆菌属(Lactobacillus,1.52%)、鞘脂单胞菌属(Sphingomonas,1.23%)(图7)。
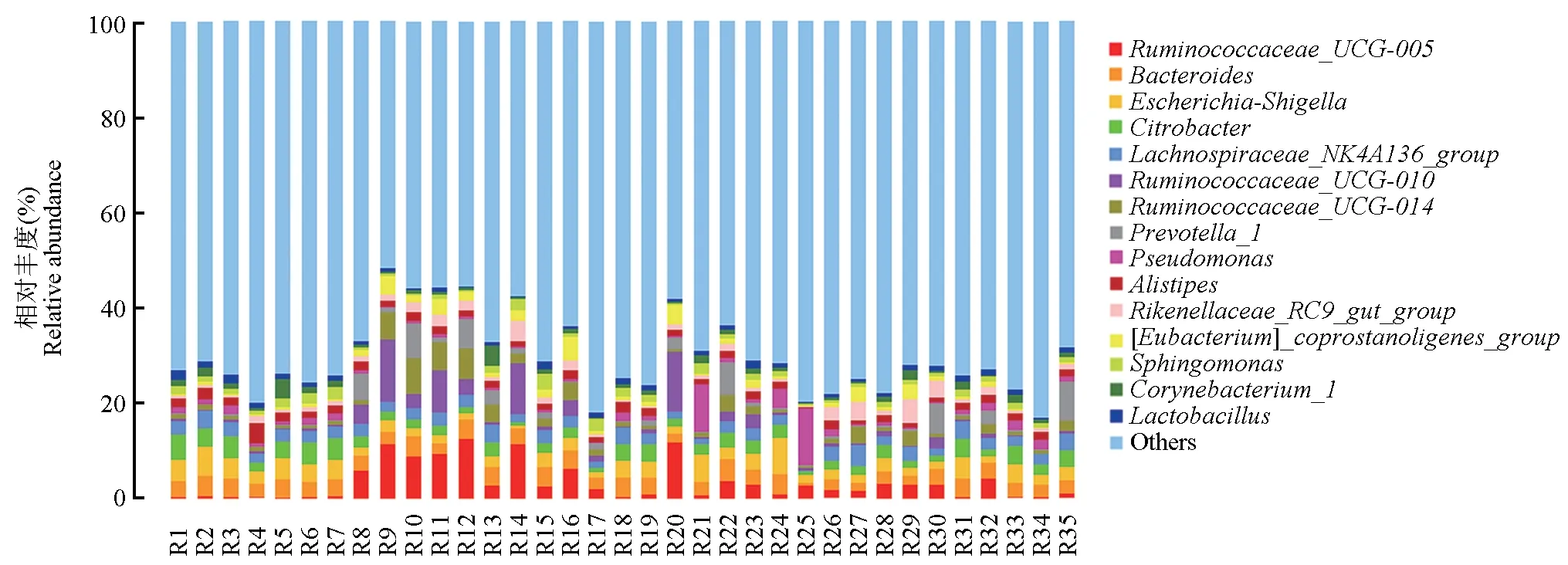
图7 马鹿肠道菌群属水平相对丰度Fig.7 Relative abundance of gut bacteria composition at the genus level
3 讨论
随着第三代测序技术愈发成熟,高通量测序技术成为研究动物肠道微生物的重要技术手段,如在大熊猫(Ailuropoda melanoleuca)[18]、黑 犀(Diceros bicornis)[19]、驼鹿(Alces alces)[20]和川金丝猴(Rhinopithecus roxellana)[21]等肠道微生物研究中均使用该技术进行测序分析。在驼鹿[20]、东北马鹿[22]和牦牛(Bos grunniens)[23]等动物肠道中,厚壁菌门和拟杆菌门都是优势菌门;在大熊猫[24]、野猪[25]中变形菌门是优势菌门。可见,不同物种之间肠道微生物存在差异,这与物种栖息地、食性、年龄、性别甚至季节都有很大关系。
本研究探究了夏季赛罕乌拉马鹿肠道微生物的多样性,除1 份样品可能存在测序深度不足外,其余样品中厚壁菌门、拟杆菌门、变形菌门和放线菌门是主要优势菌门,这与贺兰山马鹿夏季肠道微生物的研究结果[26]类似,但菌群丰度存在一定差距,赛罕乌拉马鹿的变形菌门和拟杆菌门的丰度大于贺兰山马鹿,厚壁菌门则较低。在一些有蹄类动物肠道微生物研究中,往往都是厚壁菌门占优势,拟杆菌门次之,变形菌门和其余菌门丰度较小,这可能与菌群在体内发挥的作用不同有关。厚壁菌门在动物体内往往起到降解纤维、促进消化与吸收的作用,同时厚壁菌门会促进脂肪积累和动物体重增加,但厚壁菌门中的链球菌会引起猩红热、脓疱病等[27];拟杆菌门同样可以促进食物的消化与吸收,其主要对蛋白质及碳水化合物发挥作用,在消化方面往往与厚壁菌门达到协同作用,同时拟杆菌门也会感染宿主出现炎症,如腹泻或脓肿[28];变形菌门被认为是体内的危险因素,当其丰度剧烈变化时往往会伴随疾病的发生[29],可见变形菌门是体内肠道微生物健康与否的预警者,在个体尚未出现明显症状时动物体内的变形菌群丰度就已经快速上升,食性、环境改变也会使得个体的变形菌门快速响应[30]。本研究中的菌群构成与李俊乐[26]的冬、夏季马鹿研究结果相对比,变形菌门、酸杆菌门和志贺氏菌的丰度偏高,可能与赛罕乌拉保护区马鹿感染肠炎等有关。出现肠炎的小鼠变形菌门丰度明显高于正常小鼠,肺炎克雷伯氏菌(Klebsiella pneumoniae)和奇异变形杆菌(Proteus mirabilis)等会导致小鼠体内发生不恰当的免疫反应,造成局部炎症[31]。田丽[32]在对炎症性肠病研究中也证实,炎症个体肠道菌群变形菌门、Epsilonbacteraeota 和酸杆菌门丰度升高,厚壁菌门和拟杆菌门丰度明显降低。在肠炎小鼠与正常小鼠肠道菌群研究中,在属水平上普雷沃菌属、埃希氏菌-志贺氏菌属和柠檬酸杆菌属肠炎组丰度增加,与正常组小鼠有显著区别[30]。柠檬酸杆菌中许多血清变型的抗原与埃希氏菌-志贺氏菌属的抗原有关,而普雷沃菌属也是常见的条件致病菌,由其介导的黏膜炎症可能会影响全身疾病[33];毛螺菌属和另枝菌属有促进肠黏膜修复的功能[34]。因此,赛罕乌拉保护区马鹿或许存在肠道疾病风险,具体原因还需进一步研究,但在以后保护策略制定中应重点关注。
本研究中马鹿个体肠道微生物之间分散均匀,无明显差别,这可能是食物影响造成不同个体肠道微生物趋同。在大熊猫的肠道微生物研究中发现,独特的食性使得大熊猫肠道微生物产生适应,食物的改变会使得菌群随之变化[35];在圈养马麝(Moschus chrysogaster sifanicus)和野生马麝的研究中也推测食物是造成肠道菌群差异的主要因素,圈养个体的变形菌门和拟杆菌门丰度都高于野生个体[36];有趣的是,驼鹿在夏季取食林间边缘草本植物和沼泽旁的水生植物时,体内的肠道菌群会受到取食地所处环境的影响[20],显示来自环境微生物的效应。本研究与此前对赤峰地区东北马鹿冬季肠道微生物多样性研究在群落丰度[14]上有明显区别,冬季时厚壁菌门丰度(79.43%)远大于本研究的夏季(29.30%),而拟杆菌门(冬季16.64%,夏季29.55%)和变形菌门(冬季0.40%,夏季29.30%)丰度明显减少,冬季的马鹿食物多是高纤维的木本植物,需要更多厚壁菌门参与高纤维的消化与吸收,为马鹿活动提供能量;夏季时马鹿取食植物多为草本植物,主要采食嫩芽、嫩叶等,因此拟杆菌门丰度更高。在阿拉善马鹿冬、夏肠道菌群结构研究中发现,季节对马鹿肠道菌群有重要影响,但发挥主要作用的还是食物[26]。以上研究都说明食物是动物肠道微生物群落进化的主要驱动力,肠道微生物构成反映了食物变化对动物的影响。为更加深入了解季节变化对本区域马鹿的影响以及动物的适应,建议后期开展不同季节和食物组成的对比研究,从而为提高马鹿保护管理成效提供充分的基础信息。
猜你喜欢
中国比较医学杂志(2020年4期)2020-05-26
水生生物学报(2019年4期)2019-07-20
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
小哥白尼(野生动物)(2018年1期)2018-05-26
纤维复合材料(2018年4期)2018-04-28
新疆大学学报(自然科学版)(中英文)(2016年4期)2016-05-16
石油化工建设(2016年4期)2016-02-27
大型铸锻件(2015年1期)2016-01-12
上海金属(2014年4期)2014-12-15
