
加权基因共表达网络分析挖掘老鸦瓣芽茎发育关键基因
2023-02-21 05:58张军霞郭巧生朱再标徐碧霞
中草药 2023年4期
张军霞,郭巧生,朱再标,徐碧霞
• 药材与资源 •
加权基因共表达网络分析挖掘老鸦瓣芽茎发育关键基因
张军霞,郭巧生,朱再标*,徐碧霞
南京农业大学 中药材研究所,江苏 南京 210095
利用老鸦瓣各部位及芽茎不同发育时期RNA-Seq及基因表达量数据,挖掘老鸦瓣芽茎发育过程关键基因。通过加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)法构建网络,根据模块功能富集分析及基因表达模式进行共表达模块和核心基因的筛选。通过基因表达量相关性进一步将网络划分为15个模块,将共表达模块与老鸦瓣芽茎3个发育时期相关联,鉴定到与芽茎发生高度相关的3个模块,即T1 MEplum1模块、T2 MEdarkturquoise模块和T3 MElightcyan模块。对3个模块内的基因进行动态基因本体(gene ontology,GO)和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)功能富集分析并绘制网络关系图,挖掘出4个与芽茎发育过程相关核心基因(、、、),涉及蛋白质合成、次生代谢产物的糖基化修饰、植物激素调节等。挖掘出的3个共表达模块和4个核心基因有助于进一步阐明老鸦瓣芽茎发育机制。
老鸦瓣;转录组;加权基因共表达网络分析;芽茎;富集分析;核心基因
老鸦瓣(Miq.) Honda为百合科老鸦瓣属多年生草本植物[1]。其去除膜质皮和披绒毛的干燥鳞茎为药材光慈菇,味甘、性寒,有小毒,有清热解毒、散结消肿功效,用于治疗咽喉肿痛、瘰疬、乳腺癌等多种癌症[2-3]。由于近年来光慈菇药用价值的不断开发,对老鸦瓣的需求日益增长。但目前老鸦瓣以野生资源为主,人工栽培技术还在逐步完善中。老鸦瓣人工栽培中一个重要限制因子是繁殖系数。前期研究发现在1月底2月初,生长年限较短的老鸦瓣从鳞茎盘的侧面形成1~4条地下茎状结构(暂命名“芽茎”),而芽茎是影响老鸦瓣繁殖系数的主要因素[4]。
老鸦瓣芽茎兼具根、根状茎或匍匐茎的部分特点,作为重要的无性生殖器官,对老鸦瓣快速繁育有重要作用,也可作为研究老鸦瓣物种进化和生态适应等方面的重要材料。课题组前期研究了影响老鸦瓣芽茎形成的环境条件[5],芽茎发育过程的糖代谢物质和内源激素含量变化[6],通过对老鸦瓣芽茎转录组测序,筛选到大量差异表达基因,可能参与了细胞生长、激素调节、代谢等过程并筛取部分基因进行了验证[7]。通过对芽茎发生的miRNA测序并分析,发现5个miRNA可能在芽茎发育过程具有重要作用[8]。
随着生命科学的发展,更多系统生物学的研究方法应用于植物器官发育的研究,如加权基因共表达网络分析(weighted correlation network analysis,WGCNA)[9]。WGCNA被广泛应用于医学和植物学研究领域,核心基因与其他基因连接最紧密,更容易发现与性状相关的生物学意义[10],用于鉴定候选生物标记基因或治疗靶点[11-12]。
本研究将WGCNA法与转录组测序结合,构建老鸦瓣6个组织的加权基因共表达网络并划分模块,探索老鸦瓣芽茎不同发育阶段基因表达模式并分析特异性模块,以期发掘芽茎发生的核心基因,为进一步研究调控老鸦瓣芽茎发育的分子机制提供依据。
1 材料
本实验数据来源于课题组前期对老鸦瓣不同器官及芽茎不同发育时期的转录组测序结果[7-8]。老鸦瓣于2018年10月栽种于南京农业大学试验大棚,实验材料经南京农业大学郭巧生教授鉴定为老鸦瓣(Miq.) Honda。选取其叶(L)、花冠(F)、鳞茎(B)及芽茎发育的前、中、后期(T1、T2、T3)6个时期样品,每个重复3次,共18个样品。
2 方法
2.1 WGCNA的构建流程
基于WGCNA软件包和R包进行样品聚类以及数据缺失值检测,并基于基因表达数据变异程度过滤,选择绝对中位差(median absolute deviation,MAD)前75%且MAD>0.1的基因进行后续分析。通过使用WGCNA软件包的选择软阈值的功能进行网络拓扑分析[12],选择软阈值β=21(拟合指数>0.9)进行邻矩阵的构建,建立网络,基于cutHeight=0.25(相当于模块相似的高于0.75)进行相似模块合并,最获得16个模块(其中,grey模块含未匹配到任何模块的基因),选择除grey模块之外的15个模块做分析。结合WGCNA网络、模块划分结果与样本性状联合分析模块和样本之间的关系。再通过模块内连通性(module eigengene-based connectivity,kME)找寻每个模块的核心基因,在共表达模块内具有高网络连接性的基因被定义为核心基因[13]。
2.2 特异性模块的筛选
模块与性状相关性绝对值代表着模块与性状之间的相关性。某一性状与某一模块的相关性绝对值越接近1,性状与模块的基因功能相关性越大[14]。选择绝对值大于0.6的模块作为进一步分析的组织特异性模块。
2.3 模块基因本体(gene ontology,GO)和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)功能富集分析
通过转录组数据获得基因的GO编号,利用基迪奥云平台网页https://www.omicshare.com/tools 在线分析模块的GO富集和KEGG通路富集。
2.4 核心基因的筛选与功能预测
通过分析结果中模块kME中最高的150个基因的“点”“线”关系文件,绘制基因的Cytoscape网络关系图,选定位于网络关系图中颜色较深的基因为核心基因。经NCBI的blast对筛选得到的基因进行序列比对,根据结果,初步确定其所属基因家族,预测其功能,进一步确定与老鸦瓣芽茎阶段生长发育相关的候选基因。
3 结果与分析
3.1 WGCNA的构建
通过WGCNA方法构建老鸦瓣加权基因共表达网络,筛选出具有较高表达量的39 931个Unigene参与网络划分,并进行相似模块合并,最终获得了15个共表达模块(图1),每个模块用不同的聚类树和不同颜色表示,其中,darkturquoise(暗绿松石色)模块中基因数目最多,达16 101个,palevioletred(苍紫罗蓝色)模块基因数目最少,为63个。图2分为3部分,一是基于拓扑重叠(topological overlap dissimilarity measure,TOM)计算基因间相关联程度绘制的基因系统聚类树,表现每个基因的聚类关系;二是对应第1部分结果,使用动态剪枝算法将基因划分模块的结果;三是对第2部分结果进行优化合并,获得的最终模块划分结果。

图1 基因聚类树和模块划分
3.2 组织特异性模块的分析
通过计算模块与形状相关性绝对值,发现3个组织和芽茎3个发育阶段共有15个组织特异性模块(绝对值>0.6),包括呈正相关和负相关(图2)。其中多数在芽茎T3时期、花、鳞茎、叶片。与芽茎发育后期T3正相关的模块有浅青色(MElightcyan,0.73)、蓟色(MEthistle1,0.68)模块;与鳞茎正相关的模块有黄色(MEyelow,0.90)、浅黄色(MElightyellow,0.67)、棕色4(MEbrown 4,0.76)模块;与叶片呈正相关的是棕色(MEbrown,0.98)模块;与花呈正相关的是绿色(MEgreen,0.98)、洋红色(MEmagenta,0.82)模块。与T1呈负相关的是紫红色(MEplum1,0.66)模块;与叶片负相关的模块有深橙色2(MEdarkorange 2,0.89);与花负相关的模块有夜蓝色(MEmidnightblue,0.62)。模块与组织相关性绝对值表明模块与组织的关联性,数值越大关联度越高。从中筛选的组织特异性模块参与各组织特有的生物学过程的可能性越大。为了验证基因共表达网络和组织特异性模块两者结果的可靠性,同时为了研究芽茎发育过程,对T1、T3的组织特异性模块MEplum1、MElightcyan与T2相关系数高的模块MEdarkturquoise做进一步分析。
3.3 芽茎的组织特异性表达
通过对芽茎发育T1、T2、T3时期的MEplum1、MEdarkturquoise、MElightcyan模块做GO富集分析(图3-A~C)。3个模块相比较,在生物学过程(biological progress,BP)、细胞组分(cellur component,CC)、分子功能(molecular function,MF)3个方面均发挥主要作用。其中的生物过程包括了细胞过程、代谢过程、固着生物过程,分子功能包括结合、催化活性,细胞组分包括细胞、细胞组成、细胞器等均占有较大比重。MEdarkturquoise模块基因与MEplum1模块中的基因相比,生物学过程中的细胞成分组织或生物发生、生长发育过程、生物节律过程所占比率有所上升,MElightcyan模块的定位、细胞成分组织或生物发生及生长发育过程的比重增加;在分子功能方面,MEdarkturquoise模块的基因在抗氧化活性、核苷酸结合转录因子、信号转换器所占比率增加,其中抗氧化活性提升的比例较高,MElightcyan模块则是催化活性、转运体活性的比率有所上升;在MEdarkturquoise模块基因的细胞组分方面,胞外区的比例明显增加,MElightcyan模块基因的细胞膜、细胞膜组成这2项较MEplum1模块基因的比例有增加。

图2 老鸦瓣6个组织特异性模块
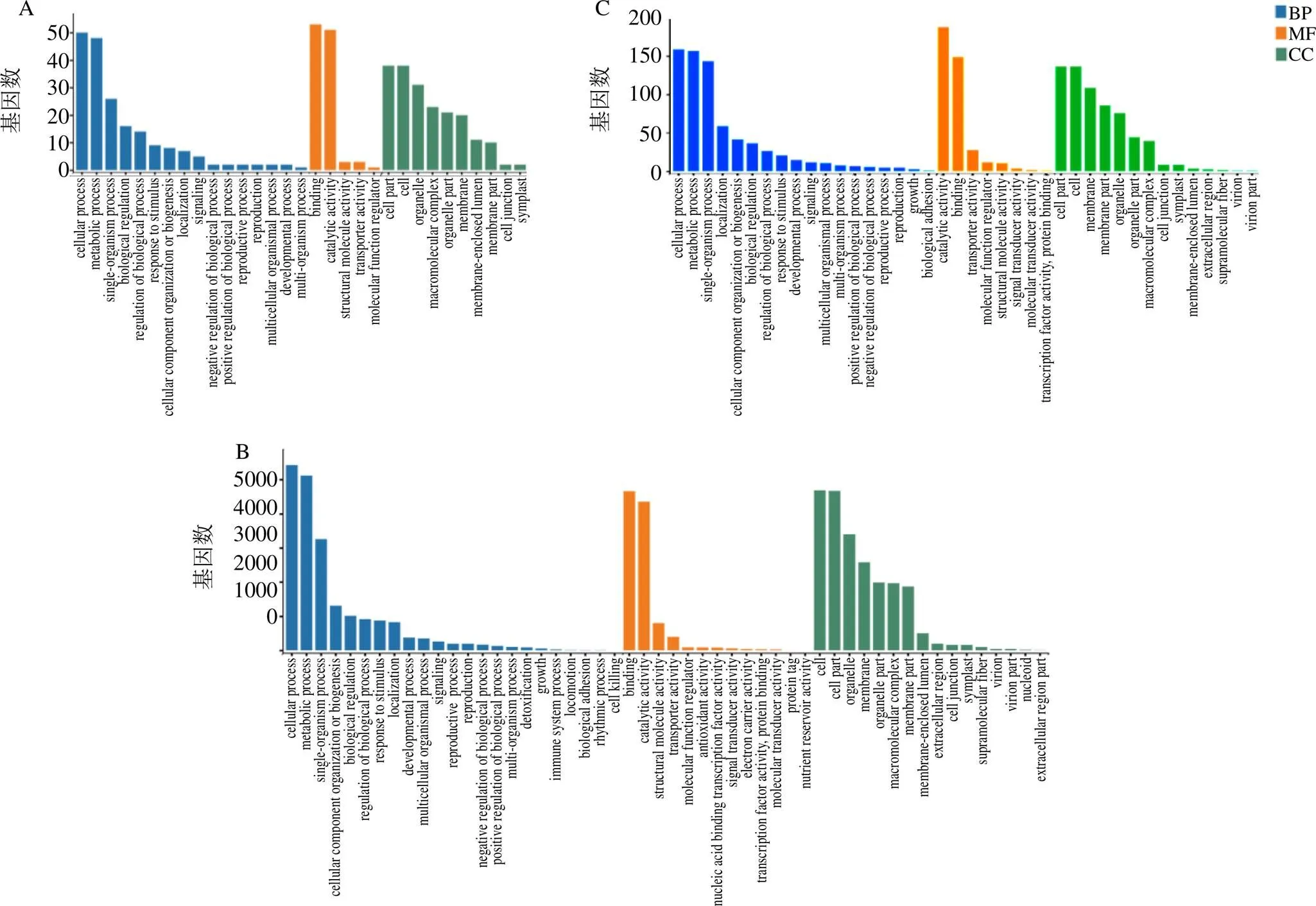
A-T1 MEplum1模块 B-T2 MEdarkturquoise模块 C-T3 MElightcyan模块
再以芽茎T1模块内的基因为对照,着重对芽茎的T2模块进行KEGG富集分析(图4-A~B),结果发现,与MEplum1模块相比,MEdarkturquoise模块的基因在代谢过程的比例略有增加,主要是在氨基酸代谢、脂类代谢、能量代谢、次生代谢及生物合成、糖类生物合成与代谢;基因信息处理过程的翻译等方面发挥着主要作用。而与MEdarkturquoise模块相比,MEplum1模块基因无明显差异。
3.4 模块核心基因的筛选与功能预测
为缩小筛选范围,选取模块kME较高的150个基因绘制网络关系图(图5-A~C),根据网络图示信息,将T2时期kME较高同时权重值大的73个基因分布在5个类别下,分别含有45、11、7、3、7条基因,将权重值最高的第一个类别中的45个基因绘制热图分析(图6),并通过BLASTP比对,发现15条序列能确切比对到信息,其中有6条比对到核糖体蛋白,找到2条已经试验验证过的核糖体蛋白序列、,同时基于本课题组前期对老鸦瓣芽茎的生理生化分析,找到注释为糖基转移酶的和注释为细胞色素P450的的4条基因序列。

A-T1 MEplum1模块 B-T2 MEdarkturquoise模块
其中,T2 MEdarkturquoise模块中的、比对到核糖体蛋白XP_013683027.1、KAE8711130.1,比对到细胞色素P450 704C1 OAY69779.1。为糖基转移酶RWR91480.1。
4 讨论
在前期研究中发现,通过转录组测序,用GO、COG、KEGG数据库对芽茎形成发育的差异基因进行注释,功能注释显示这些差异基因主要涉及物质及能量代谢、激素信号、细胞生长、转录调控等诸多生理生化过程[7]。蔗糖等可溶性糖及淀粉在众多酶,包括蔗糖磷酸合酶(sucrose phosphate synthase,SPS)、淀粉酶(amylase,AMY)、蔗糖合酶(sucrose synthase,SS)、腺苷二磷酸葡萄糖焦磷酸化酶(adenosine diphosphoglucose pyrophosphorylase,AGPase)、可溶性淀粉合成酶(soluble starch synthase,SSS)、颗粒结合型淀粉合成酶(granule-bound starch synthase,GBSS)的共同作用下大量积累,为芽茎发生时的细胞分裂和生长提供物质和能量需要;赤霉素(gibberellins,GA)、玉米素核苷(zeatin riboside,ZR)、吲哚-3-乙酸(indole-3-acetic acid,IAA)及脱落酸(abscisic acid,ABA)协同作用,相互制约平衡,共同作用于芽茎的发生[6]。
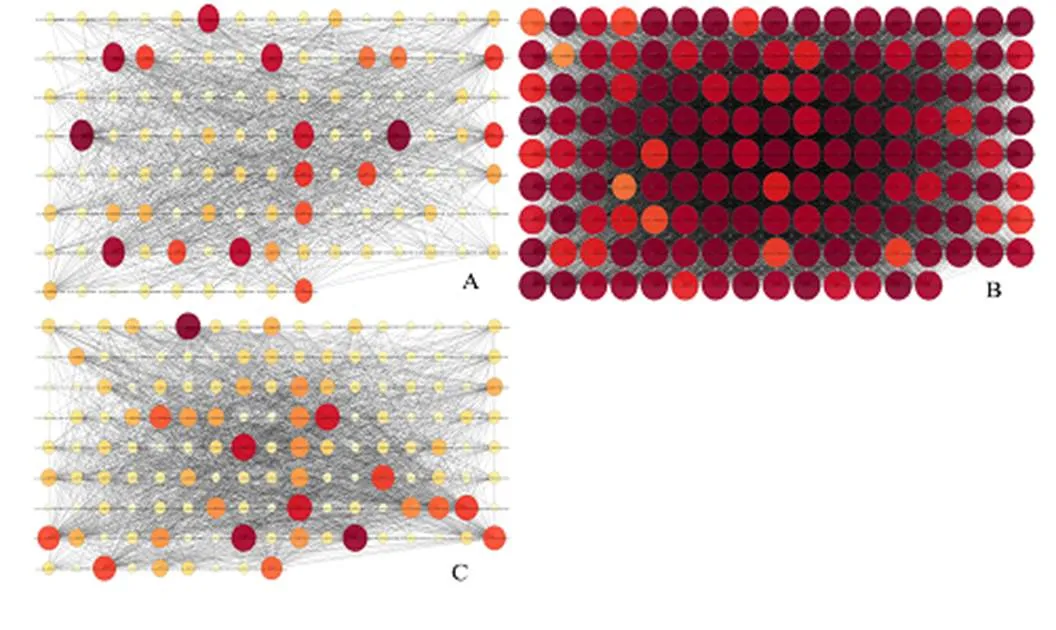
A-T1 MEplum1模块基因网络 B-T2 MEdarkturquoise模块基因网络 C-T3 MElightcyan模块基因网络
箭头代表筛选的4个基因
WGCNA能特异地筛选出与目标性状相关的基因,通过进行模块化分类,得到相关度高的共表达模块,已经被证明是一种高效的数据挖掘手段[15]。Hollender等[16]利用WGCNA法发现了草莓中与花托组织特异性有关的模块,并挖掘出7个相关核心基因。王思瑶等[14]通过WGCNA法构建网络,对特异性模块进行分析,发掘出36个潜在的与偃松种子萌发相关的核心基因。鞠正等[17]利用番茄组织的RNA-Seq通过WGCNA法构建网络分析,在网络和组织特异性模块发现了与果实成熟相关的转录因子。
本研究利用老鸦瓣芽茎与其他组织转录组数据,通过WGCNA法构建了15个基因共表达模块,分析3个芽茎发育相关的组织模块,分别为T1 MEplum1、T2 MEdarkturquoise、T3 MElightcyan模块,同时对模块进行GO、KEGG富集分析并绘制模块内基因的网络分析图,于T2 MEdarkturquoise模块筛选出4个核心基因,其可能在芽茎发育过程起到重要作用。
筛选出的4个核心基因中,、比对到核糖体蛋白。核糖体蛋白多存在于快速增殖、分泌或功能旺盛的细胞中,并参与植物发育过程[18]。除了与核糖体RNA组装形成核糖体参与细胞内蛋白质合成外,还与DNA复制、RNA加工、细胞增殖、生长发育等调控有关[19]。比对到糖基转移酶,以家族形式存在,在次生代谢产物的糖基化修饰、内源或外源物质的解毒、机体防御反应、植物激素调节等方面发挥着重要作用[20]。拟南芥中新型糖基转移酶基因能对生长素的主要前体吲哚丙酮酸(IPyA)进行特异糖基化修饰,对活性生长素本身却没有活性。通过分析激素水平,发现UGT76F1催化的IPyA糖基化能够调控IAA生物合成的代谢,参与生长素稳态调节[21]。同时,糖基转移酶家族在糖类代谢过程中参与糖类转移,其中经催化活化的木糖基转移酶是多糖生物合成中的关键酶[22]。比对到细胞色素P450,是一种氧化酶家族,广泛参与植物代谢过程,具有催化活性,主要涉及植物激素、生物碱、萜类等代谢产物的合成[23]。糖基转移酶基因为金铁索的三萜皂苷类化合物合成修饰环节的关键基因,与P450单加氧酶基因对三萜皂苷类化合物的共同母核——β-香树素进行修饰,最终形成三萜皂苷[24]。
Miao等[6]研究发现老鸦瓣的芽茎由发生T1时期至膨大T3时期到成为新鳞茎的过程中,淀粉和蛋白质的含量持续上升,可溶性糖被大量消耗,含量呈下降趋势。同时,生长素的含量在T1时期达到顶峰,于T2时期维持较高水平,T3时期显著下降。由此推测在芽茎发生过程中可能有大量的核糖体蛋白参与蛋白质的合成,且高含量生长素促进芽茎膨大,在此过程中核糖体蛋白的活性最高,通过能量合成大量芽茎发育膨大所需的蛋白质,为此提供了物质基础。
老鸦瓣芽茎膨大后形成的鳞茎入药,其中含有皂苷类、多糖类成分,推测、基因可能为老鸦瓣的次生代谢研究提供物质基础。
本研究重点关注了与老鸦瓣芽茎发育相关模块,通过WGCNA法的模块组织相关性分析挖掘出潜在的与芽茎发育相关的4个核心基因,为进一步探究核心基因与芽茎发育的关系奠定了坚实基础,后续将对这些基因的功能进行验证。
利益冲突 所有作者均声明不存在利益冲突
[1] 谭敦炎, 张震, 李新蓉, 等. 老鸦瓣属(百合科)的恢复: 以形态性状的分支分析为依据 [J]. 植物分类学报, 2005, 43(3): 262-270.
[2] 国家中医药管理局《中华本草》编委会. 中华本草: 藏药卷 [M]. 上海: 上海科学技术出版社, 2002: 176-177.
[3] 陈彪, 焦淑萍, 尹荣, 等. 6种吉林抗癌中药清除羟自由基及其抗DNA损伤体外实验研究 [J]. 第三军医大学学报, 2004, 26(1): 88-89.
[4] 杨小花, 缪媛媛, 郭巧生, 等. 不同等级老鸦瓣生长繁殖特征研究 [J]. 中草药, 2015, 46(24): 3746-3750.
[5] 缪媛媛. 老鸦瓣芽茎发育特征及形成机制研究 [D]. 南京: 南京农业大学, 2015.
[6] Miao Y Y, Zhu Z B, Guo Q S,. Dynamic changes in carbohydrate metabolism and endogenous hormones duringstolon development into a new bulb [J]., 2016, 59(2): 121-132.
[7] Miao Y Y, Zhu Z B, Guo Q S,. Transcriptome analysis of differentially expressed genes provides insight into stolon formation in[J]., 2016, 7: 409.
[8] Zhu Z B, Miao Y Y, Guo Q S,. Identification of miRNAs involved in stolon formation inby high-throughput sequencing [J]., 2016, 7: 852.
[9] Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis [J]., 2008, 9: 559.
[10] 刘源, 隋正红, 刘昊昕, 等. 加权基因共表达网络探究链状亚历山大藻爆发性生长的分子机制 [J]. 中国海洋大学学报: 自然科学版, 2019, 49(9): 66-76.
[11] Yin L, Cai Z H, Zhu B A,. Identification of key pathways and genes in the dynamic progression of HCC based on WGCNA [J].(), 2018, 9(2): E92.
[12] Gao C, Ju Z, Li S,. Deciphering ascorbic acid regulatory pathways in ripening tomato fruit using a weighted gene correlation network analysis approach [J]., 2013, 55(11): 1080-1091.
[13] Fuller T F, Ghazalpour A, Aten J E,. Weighted gene coexpression network analysis strategies applied to mouse weight [J]., 2007, 18(6/7): 463-472.
[14] 王思瑶, 丛日征, 闫晓娜, 等. 利用加权基因共表达网络分析 (WGCNA) 的方法挖掘偃松种子萌发过程关键基因 [J]. 温带林业研究, 2019, 2(1): 39-46.
[15] Zhao W, Langfelder P, Fuller T,. Weighted gene coexpression network analysis: State of the art [J]., 2010, 20(2): 281-300.
[16] Hollender C A, Kang C Y, Darwish O,. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks [J]., 2014, 165(3): 1062-1075.
[17] 鞠正, 曹东艳, 梁岩, 等. 利用加权基因共表达网络分析 (WGCNA) 的方法挖掘番茄果实成熟相关的转录因子 [J]. 中国食品学报, 2018, 18(6): 240-248.
[18] 廖昌敏, 张咏祀, 刘小红, 等. 水杉40S核糖体蛋白S8基因的克隆及生物信息学分析 [J]. 分子植物育种, 2020, 18(21): 7008-7014.
[19] Labriet A, Lévesque É, Cecchin E,. Germline variability and tumor expression level of ribosomal protein gene RPL28 are associated with survival of metastatic colorectal cancer patients [J]., 2019, 9(1): 13008.
[20] 马风伟, 邓青芳, 陈海江, 等. 植物UDP-糖基转移酶结构及活性研究进展 [J]. 贵阳学院学报: 自然科学版, 2019, 14(3): 73-82.
[21] Chen L, Huang X X, Zhao S M,. IPyA glucosylation mediates light and temperature signaling to regulate auxin-dependent hypocotyl elongation in[J]., 2020, 117(12): 6910-6917.
[22] Han W T, Fan X, Teng L H,. Identification, classification, and evolution of putative xylosyltransferases from algae [J]., 2019, 256(4): 1119-1132.
[23] 朱灵英, 郭娟, 张爱丽, 等. 参与植物三萜生物合成的细胞色素P450酶研究进展 [J]. 中草药, 2019, 50(22): 5597-5610.
[24] 江舟. 金铁锁糖基转移酶的克隆与生物信息学分析 [D]. 昆明: 云南中医学院, 2016.
Key Genes involving in stolon development ofwith weighted gene co-expression network analysis
ZHANG Jun-xia, GUO Qiao-sheng, ZHU Zai-biao, XU Bi-xia
Institute of Chinese Medicinal Materials, Nanjing Agricultural University, Nanjing 210095, China
To detect the hub genes of the stolon development process inby using the RNA-Seq and gene expression data from various parts ofand bud stems at different developmental periods.The network was constructed by weighted gene co-expression network analysis (WGCNA) method, and co-expression modules and core genes were screened based on module functional enrichment analysis and gene expression patterns.Fifteen modules were divided through the correlation of gene expression, the co-expression module was associated with the three developmental stages ofstolon, and three modules were identified to be highly related to stolon development, namely T1 MEplum1 module, T2 MEdarkturquoise module and T3 MElightcyan module, respectively. The analysis of gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) function enrichment of the three modules and network relationship diagrams found that four hub genes related to the stolon development process, namely,,,, which were involved in protein synthesis, glycosylation modification of secondary metabolites, plant hormone regulation etc.The identification of three co-expression modules and four hub genes mined will be helpful for further elucidating the mechanism aboutstolon development.
(Miq.) Honda; transcriptome; weighted gene co-expression network analysis; stolon; enrichment analysis; hub genes
R282.12
A
0253 - 2670(2023)04 - 1228 - 08
10.7501/j.issn.0253-2670.2023.04.023
2022-08-20
国家自然科学基金资助项目(81773834);中央高校基本科研业务费项目(KYZZ2022004)
张军霞,硕士研究生,研究方向为药用植(动)物种植(养殖)理论与技术。E-mail: 2017104127@njau.edu.cn
朱再标,教授,研究方向为药用植(动)物种植(养殖)理论与技术。Tel: (025)84395980 E-mail: zhuzaibiao@njau.edu.cn
[责任编辑 时圣明]
猜你喜欢
军事文摘(2022年24期)2022-12-30
安徽农业科学(2022年1期)2022-02-14
昆明医科大学学报(2020年12期)2021-01-26
中国生殖健康(2020年7期)2021-01-18
野草(2020年4期)2020-08-13
江汉大学学报(自然科学版)(2019年6期)2019-12-06
中华家教(2018年7期)2018-08-01
北方音乐(2017年5期)2017-04-13
中国学术期刊文摘(2016年1期)2016-02-13
中国学术期刊文摘(2016年1期)2016-02-13
