
赛乐西帕中甲磺酸酯杂质的来源分析与检测
2023-02-14 11:00马天华赖广健华莹黄雅婷刘恩萍王云中
当代化工研究 2023年2期
*马天华 赖广健 华莹 黄雅婷 刘恩萍 王云中
(普济生物科技(台州)有限公司 浙江 318000)
前言
磺酸酯类杂质作为遗传毒性杂质之一,格外受到关注。2000年欧洲药典指出原料药和磺酸在醇溶液中成盐可能产生磺酸酯类杂质,其形成机制于2006年由Snodin[1]首次预测,后来由Teasdale等通过实验证实。2007年7月6日欧洲药监局宣布抗艾滋病药物奈非那韦甲磺酸盐停止使用,原因是奈非那韦甲磺酸盐中EMS在药物剂量2.5g/d下摄入量高达2.5mg/d,远高于毒理学关注阈值,甲磺酸等磺酸类物质与微量的低级醇会反应生成烷基磺酸如甲磺酸甲酯(MMS)、甲磺酸乙酯(EMS)和甲磺酸异丙酯(IMS),它们易与DNA发生烷基化反应,从而可能成为引发癌症的诱因。因此,评估药物中磺酸酯潜在来源,对它们进行分析检测,制定合理的控制策略是非常重要和有意义的。
1.甲磺酸酯来源分析
根据文献[2-4]报道,赛乐西帕常见合成策略如图1所示。
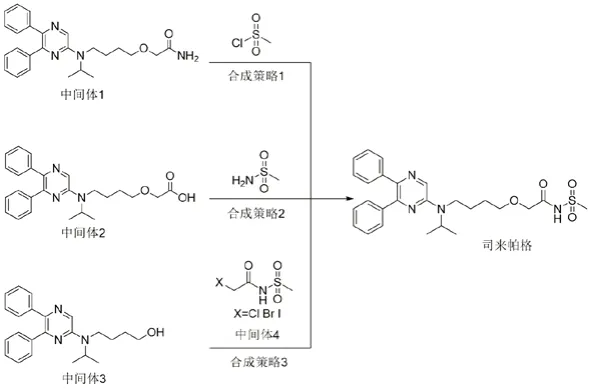
图1 赛乐西帕常见合成策略
合成策略1中使用到甲磺酰氯,采用本策略合成赛乐西帕极有可能有甲磺酸或者甲磺酰氯残留;合成策略2中使用到的甲磺酰胺,由甲磺酰氯和氨反应产生;合成策略3中的中间体4,主要由卤代乙酰卤和甲磺酰胺反应来制备,因此采用策略2和3合成赛乐西帕也有甲磺酸或者甲磺酰氯存在可能。另一方面,甲醇、乙醇、异丙醇等低级醇在原料药合成过程中使用极其普遍,而且其潜在来源如酯类溶剂的水解、醚的水解等也很常见。当甲磺酸或者甲磺酰氯在有甲醇、乙醇、异丙醇等低级醇存在时都容易发生反应生成具有遗传毒性杂质MMS、EMS和IMS。而且在这些合成策略中,甲磺酸和甲磺酰氯残留集中在合成工艺的末端,进一步增加了原料药中磺酸酯残留的风险。因此,需要基于赛乐西帕合成工艺中甲醇、乙醇和异丙醇等低级醇的实际使用情况以及工艺中低级醇潜在来源对赛乐西帕合成工艺中产生MMS、EMS和IMS的可能性进行评估。一旦确认存在甲磺酸酯的可能,应基于国际人用药品注册技术协调会(ICH)相关指导原则,结合赛乐西帕产品特性制定合理甲磺酸酯控制策略,并使用经过验证的分析方法对产品进行检测,以证明产品中甲磺酸酯含量水平是安全可控的。
2.检测方法研究进展
甲磺酸酯类杂质检测方法研究较多,常用的有气相色谱法、气相色谱-质谱联用(GC-MS)法、液相色谱-质谱联用(LC-MS)法等。例如:Taylor 等[5]使用LC-MS法在单离子模式下直接检测MMS、EMS和IMS。Alzaga等[6]报道衍生化HS-GC-MS法测定原料药中的9种磺酸酯,该方法衍生物挥发性好,检测灵敏度高,确定了磺酸酯检测通过分析物衍生的可行性。Filby等[7]利用气相色谱-火焰离子化检测器(GC-FID)对包括MMS等13种有机硫化合物进行检测。Ramjit等[8]开发了一种GC-MS方法来识别和量化正性肌力药物DPI 201-106的双甲磺酸盐中甲磺酸酯。研究结论表明DPI 201-106的游离碱中不含任何可检测水平的甲磺酸酯,而双甲磺酸盐分别含有0.51×10-6和1.31×10-6的MMS和EMS,这进一步说明甲磺酸的存在易与低级醇生成甲磺酸酯。在常用磺酸酯检测方法中,直接进样方式的气相色谱法易引入非挥发性物质,存在进样重复性差和准确度不达标;顶空进样方式的气相色谱法可以避免药品基质的干扰,但由于磺酸酯沸点较高,同样会限制方法的灵敏度;其他检测方法如LC-MS亦有基质干扰,灵敏度低等缺点。因此,根据原料药性质和磺酸酯种类不同,针对特定原料药中的磺酸酯开发检测方法是必要的。
3.检测方法开发
鉴于赛乐西帕化学性质,本试验中采用衍生化HS-GC-MS法对赛乐西帕原料药中MMS、EMS和IMS进行测定,研究方法具体如下:
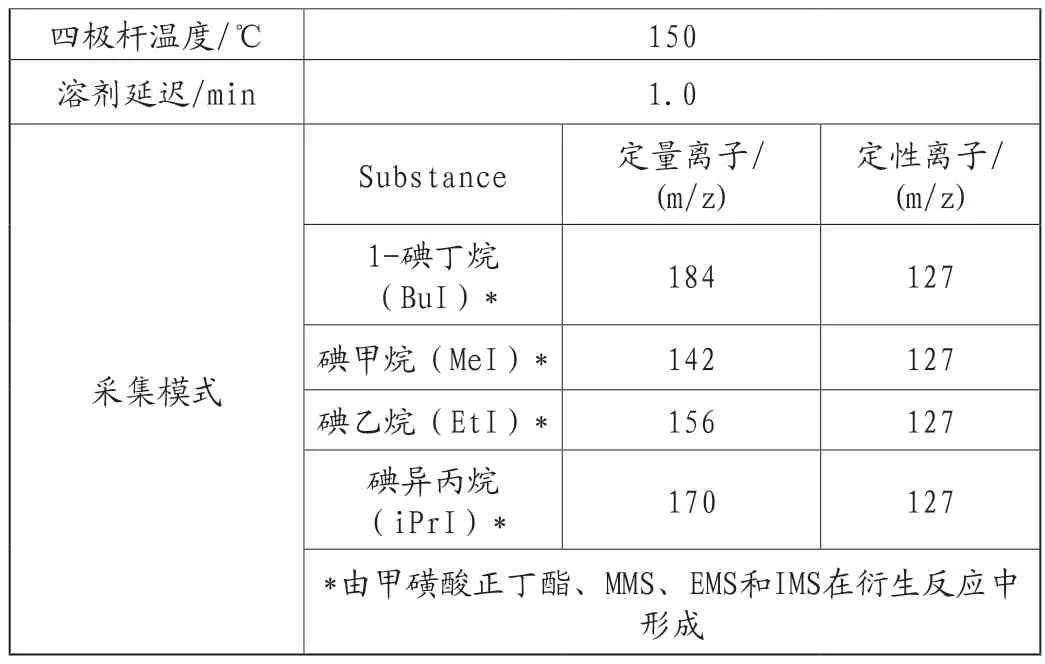
(1)色谱条件

表1 色谱条件及质谱参数
(2)溶液配制
称取碳酸氢钠5g、30mg无水硫代硫酸钠和60g碘化钠至50mL容量瓶中,加水超声溶解,并稀释到刻度得到衍生化溶液;精密移取甲磺酸正丁酯10μL,置25mL量瓶中,加甲苯稀释至刻度,再精密量取20μL,置100mL量瓶中,用DMF稀释至刻度得内标溶液;精密移取衍生化溶液0.5mL和内标溶液1.5mL,置20mL顶空瓶中,密封得到空白溶液;精密称取供试品约25mg,置于20mL顶空瓶中,精密加入衍生化溶液0.5mL和内标溶液1.5mL,密封得到供试品溶液;分别精密称取MMS、EMS和IMS各约25mg,用DMF溶解并稀释至5.0mL。精密移取50μL,置25ml容量瓶中,加内标溶液稀释至刻度,混匀得到对照品储备液,精密量取对照品贮备液0.5mL,置20mL容量瓶中,用内标溶液稀释至刻度得对照品中间储备液,精密移取0.5mL对照品中间储备液和衍生化溶液1.5mL,置20mL顶空瓶中,密封得对照品溶液。室温条件下,对照品溶液4h稳定,其方法对照品限度为5×10-6,质量浓度为0.25μg·mL-1。
4.方法学验证
(1)专属性试验
称取供试品约25mg,移取衍生试剂与对照品贮备液各1mL置顶空瓶,进样。
图2中MMS、EMS和IMS的保留时间分别为2.39min、2.84min和3.06min,各杂质峰间的分离度良好,且空白不干扰测定。

图2 专属性溶液色谱图
(2)检测限和定量限
取MMS、EMS和IMS溶液贮备液逐级稀释。信噪比为10时,MMS、EMS和IMS的定量限分为0.0127μg·mL-1、0.0136μg·mL-1和0.0125μg·mL-1;信噪比为3时,MMS、EMS和IMS的定量限分为0.0064μg·mL-1、0.0078μg·mL-1和0.0063μg·mL-1,检测限(相对供试品浓度12.5mg·mL-1)分别为1×10-6、1×10-6和1×10-6。
(3)线性考察
取MMS、EMS和IMS适量,精密称定,依次稀释,制成浓度范围在0.013~0.32μg·mL-1的系列标准溶液。采用顶空气相色谱质谱法进行检测分析,记录色谱图,以浓度为横坐标,峰面积为纵坐标,进行线性回归,并及时回归方程和相关系数,结果见表2~表4。

表2 甲磺酸甲酯的衍生化产物碘甲烷的线性与范围实验结果

表3 甲磺酸乙酯的衍生化产物碘乙烷的线性与范围实验结果
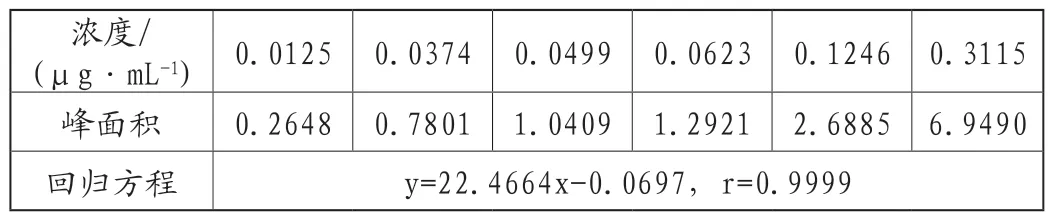
表4 甲磺酸异丙酯的衍生化产物碘异丙烷的线性与范围实验结果
(4)重复性试验
取供试品25mg,加入MMS、EMS和IMS适量,使供试品溶液中含有约0.0625μg·mL-1的MMS、EMS和IMS,即重复性溶液(n=6)。结果MMS、EMS和IMS的平均回收率分别为103.5923%、102.6765%和104.2078%,且RSD分别为1.05%、0.51%和0.38%,重复性良好。
(5)回收率试验
取供试品25mg,分别加入MMS、EMS和IMS适量,使供试品溶液中含有约0.0125μg·mL-1、0.0625 μg·mL-1和0.125μg·mL-1的MMS、EMS和IMS,即加样回收溶液(n=3)。结果MMS、EMS和IMS的平均回收率分别为105.4283%、103.0952%和103.7930%,其RSD(n=9)分别为2.85%、1.20%和0.73%,回收率良好。
(6)稳定性
取赛乐西帕供试品,加入对照溶液,即得0.0625 μg·mL-1的加标供试品溶液。分别于0h、4h、12h注入色谱仪,MMS、EMS和IMS 12h的峰面积相对于0h的峰面积未有超过5%以上的变化,考察结果表明:样品溶液稳定性良好。
(7)样品中甲磺酸酯测定
取赛乐西帕原料药各适量,按上述验证后的分析方法检测,结果显示一批有EMS检出,含量小于5×10-6,未检出MMS和IMS,另一批均未检出MMS、EMS和IMS。
5.结论
本文从不同赛乐西帕合成策略上分析赛乐西帕合成过程中产生MMS、EMS和IMS的潜在可能,开发了衍生化HS-GC-MS方法测定赛乐西帕原料药中MMS、EMS和IMS含量,并完成方法验证。该方法灵敏度高、重复性好,满足检测要求。通过对赛乐西帕样品检测,证实赛乐西帕合成过程中确实存在产生MMS、EMS和IMS并最终在API中残留的可能性。
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
分析化学(2019年3期)2019-03-30
科技与创新(2016年5期)2016-03-17
化工生产与技术(2016年5期)2016-03-13
山东工业技术(2015年6期)2015-07-27
中国生化药物杂志(2015年4期)2015-07-07
中国当代医药(2015年9期)2015-03-01
中华皮肤科杂志(2014年4期)2014-12-19
应用化工(2014年7期)2014-08-09
中国药业(2014年21期)2014-05-26
