
Suppressing high mobility group box-1 release alleviates morphine tolerance via the adenosine 5'-monophosphate-activated protein kinase/heme oxygenase-1 pathway
2023-02-13 12:41:40TongTongLinChunYiJiangLeiShengLiWanWenFanJinCanLiXiaoDiSunChenJieXuLiangHuXueFengWuYuanHanWenTaoLiuYinBingPan
中国神经再生研究(英文版) 2023年9期
Tong-Tong Lin , Chun-Yi Jiang , Lei Sheng , Li Wan Wen Fan Jin-Can Li Xiao-Di Sun, Chen-Jie Xu, Liang Hu Xue-Feng Wu, Yuan Han, Wen-Tao Liu , Yin-Bing Pan,
Abstract Opioids, such as morphine, are the most potent drugs used to treat pain. Long-term use results in high tolerance to morphine. High mobility group box-1 (HMGB1) has been shown to participate in neuropathic or inflammatory pain, but its role in morphine tolerance is unclear. In this study, we established rat and mouse models of morphine tolerance by intrathecal injection of morphine for 7 consecutive days. We found that morphine induced rat spinal cord neurons to release a large amount of HMGB1. HMGB1 regulated nuclear factor κB p65 phosphorylation and interleukin-1β production by increasing Toll-like receptor 4 receptor expression in microglia, thereby inducing morphine tolerance. Glycyrrhizin, an HMGB1 inhibitor, markedly attenuated chronic morphine tolerance in the mouse model. Finally, compound C (adenosine 5′-monophosphate-activated protein kinase inhibitor) and zinc protoporphyrin (heme oxygenase-1 inhibitor) alleviated the morphine-induced release of HMGB1 and reduced nuclear factor κB p65 phosphorylation and interleukin-1β production in a mouse model of morphine tolerance and an SH-SY5Y cell model of morphine tolerance, and alleviated morphine tolerance in the mouse model. These findings suggest that morphine induces HMGB1 release via the adenosine 5′-monophosphate-activated protein kinase/heme oxygenase-1 signaling pathway, and that inhibiting this signaling pathway can effectively reduce morphine tolerance.
Key Words: adenosine 5′-monophosphate-activated protein kinase; heme oxygenase-1; high mobility group box-1; interleukin-1β; microglia; morphine tolerance; neuroinflammation; neuron; nuclear factor-κB p65; Toll-like receptor 4 1Jiangsu Key Laboratory of Neurodegeneration, Department of Pharmacology, Nanjing Medical University, Nanjing, Jiangsu Province, China; 2Department of Anesthesiology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu Province, China; 3Department of Anesthesiology and Pain, Nanjing First Hospital, Nanjing Medical University, Nanjing, Jiangsu Province, China; 4State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing, Jiangsu Province, China; 5Department of Anesthesiology, Eye & ENT Hospital, Fudan University, Shanghai, China; 6Institute of Translational Medicine, Nanjing Medical University, Nanjing, Jiangsu Province, China
Introduction
Opioids, such as morphine, are the most potent drugs utilized to control severe pain (Lavand’homme and Steyaert, 2017). Long-term morphine use results in addiction and tolerance, which significantly hinder its application. Tolerance is defined as decreased efficacy of the opioid and a need for higher and more frequent doses to achieve the same analgesic effect (Morgan and Christie, 2011). Numerous studies over the past decade have been devoted to exploring the mechanisms underlying opioid tolerance (Liu et al., 2019; Mercadante et al., 2019).
Regarding the neurobiological mechanisms of morphine tolerance, neuroinflammation is considered to be critical (Zhao et al., 2010; Liu et al., 2019). Microglia play a major role in neuroinflammation in the central nervous system (Zhang et al., 2021; Chen et al., 2022). Persistent activation of the dorsal horn caused by the release of damage-associated molecular patterns (DAMPs) from neurons stimulates microglia to release inflammatory mediators (Schwaller and Fitzgerald, 2014). Toll-like receptor 4 (TLR4), which is predominantly expressed in microglia, is considered to be one of the most critical signaling receptors involved in microglial activation (Bettoni et al., 2008; Mizobuchi and Soma, 2021). TLR4 is one of the most well-studied of the patternrecognition receptors, which recognize specific DAMPs and subsequently induce an inflammatory response (Kumar et al., 2009). It has been reported that morphine triggers TLR4-mediated neuroinflammation by binding to MD-2, a TLR4 accessory protein (Hutchinson et al., 2010). Blocking TLR4 suppressed microglial activation and alleviated morphine tolerance, suggesting that TLR4 activation contributes to morphine tolerance (Eidson and Murphy, 2013). Heat shock proteins, high-mobility group box 1 (HMGB1), heparan sulfate (Akbarshahi et al., 2011), and fibrinogen (Wang et al., 2018) are all endogenous TLR4 agonists. Our previous study found that morphine induced the release of heat shock protein 70 from SH-SY5Y cells (Qu et al., 2017). As an important DAMP, heat shock protein 70 induces neuroinflammation through the TLR4-NOD-like receptor thermal protein domain associated protein 3 (NLRP3) signaling pathway (Qu et al., 2017). Furthermore, disulfide HMGB1 can mimic the effects of morphine-induced persistent sensitization of the TLR4 receptor (Grace et al., 2016). The released extracellular HMGB1 increases the release of proinflammatory cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) (Andersson and Tracey, 2011). HMGB1, which is a non-histone DNA-binding protein, is mainly located in the cell nucleus, and can stabilize nucleosome formation, as well as regulating interactions between transcription factors and DNA under physiological conditions (Angelopoulou et al., 2018). A significant amount of HMGB1 is released from the nucleus into the extracellular space immediately after neuronal injury (Paudel et al., 2018). In addition, HMGB1 is involved in neuroinflammation, modulates autophagy and apoptosis, and regulates gene transcription (Wan et al., 2016; Foglio et al., 2019). It is considered to be a critical factor in several pathophysiological conditions. Active HMGB1 secretion from inflammatory cells and passive HMGB1 release from dying neurons influence cell viability and apoptosis, and contribute to the neuroinflammation and neurodegeneration associated with Parkinson’s disease (Angelopoulou et al., 2018). HMGB1 release from astrocytes mediated by a pannexin-1 and P2X7 receptor signaling cascade can contribute to the development of major depressive disorder (Hisaoka-Nakashima et al., 2020). Qian et al. (2020) reported that chronic morphine exposure resulted in an increase in HMGB1 expression in the dorsal horn of the spinal cord. HMGB1 upregulation induced by any of these pathways contributes to the development and maintenance of morphine tolerance and hyperalgesia via TLR4-nuclear factor kappa-B (NF-κB) signaling (Qian et al., 2020).
Taken together, these earlier studies suggest that HMGB1 may be released from neurons in response to chronic morphine administration, and that extracellular HMGB1 could be a critical morphine-induced inflammatory trigger that contributes to morphine tolerance. Here, we investigated the role of HMGB1 in the pathogenesis of morphine tolerance, as well as its underlying mechanisms.
Methods
Animals
Adult male Institute of Cancer Research (ICR) mice (8 weeks old, 18–22 g) and adult male Sprague-Dawley rats (200–250 g) were provided by the Experimental Animal Center at Nanjing Medical University, Nanjing, China (license No. SCXK (Su) 2019-0001). Animals were housed five to six per cage under pathogen-free conditions with soft bedding under standard conditions (temperature 22 ± 2˚C, humidity 50 ± 5%) and a 12/12-hour light/dark cycle (lights on at 8:00 a.m.). Behavioral testing was performed during the light cycle (between 9:00 a.m. and 5:00 p.m.). The animals were allowed to acclimate to these conditions for at least 2 days before the experiments began. For each group of experiments, the animals were matched by age and body weight. Rats were randomly divided into vehicle and morphine groups (n= 6/group). In order to investigate the effect of HMGB1 inhibition on morphine tolerance, mice were randomly divided into vehicle, morphine, morphine + glycyrrhizin, and glycyrrhizin groups (n= 8/group). In order to investigate the effect of AMPK inhibition on morphine tolerance, mice were randomly divided into vehicle, morphine, morphine + Compound C, and Compound C groups (n= 8/group). In order to investigate the role of microglia in morphine tolerance, mice were randomly divided into vehicle, morphine, morphine + PLX3397 (could induce a strong decrease in microglia), and PLX3397 groups (n= 5/group). All procedures were performed strictly in accordance with the regulations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8thed, National Research Council, 2011). All animal experiments were approved by the Nanjing Medical University Animal Care and Use Committee (No. IACUC-2005001) on March 18, 2020.
Tolerance model and behavioral analysis
The animals were habituated to the testing environments for 2 days, and behavioral testing was carried out in a blinded manner. For the chronic tolerance test, mice or rats were intrathecally injected with normal saline (10 µL) or morphine (10 µg/10 µL, Shenyang First Pharmaceutical Factory, Northeast Pharmaceutical Group Company, Shenyang, China) once daily for 7 consecutive days. To administer the intrathecal injections, the mice were placed in a prone position, and the midpoint between the tips of the iliac crest was located. A Hamilton syringe with a 30-gauge needle was inserted into the subarachnoid space of the spinal cord between the L5 and L6 spinous processes.
Behavioral testing was performed by tail-flick assay 30 minutes after morphine administration every morning (Cecchi et al., 2008). The tail-flick test was performed using a water bath maintained at 52°C. Each animal was gently wrapped in cloth during the experiment. The distal one-third of the tail was immersed in the water bath, and the mice rapidly removed their tails from the bath at the first sign of discomfort. The chronometer was stopped as soon as the mouse withdrew its tail from the hot water, and the latency time was recorded. A cut-off time of 10 seconds was set to avoid tissue damage. Intrathecal injection did not affect baseline responses (latencies recorded before injection).Different doses of glycyrrhizin (25, 50, 100 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) were administered by intragastric injection 15 minutes before morphine administration once a day for 7 consecutive days. Different doses of Compound C (1, 3, or 10 µg/10 µL, MedChemExpress, Monmouth Junction, NJ, USA) were administered by intrathecal injection 15 minutes before morphine administration once a day for 7 days. On day 1, the tail-flick test was performed at 30, 60, 90, 120, 150, and 180 minutes after morphine administration to investigate whether glycyrrhizin or Compound C enhanced the acute analgesic effect of morphine. The tail-flick test was then performed once a day for 7 consecutive days after morphine administration to investigate whether glycyrrhizin or Compound C enhanced the chronic analgesic effect of morphine.
Mice received PLX3397 (40 mg/kg, MedChemExpress) intragastrically once a day for 10 days in total starting 3 days before morphine administration. The tail-flick test was performed for 7 consecutive days after morphine administration to investigate whether PLX3397 enhanced the chronic analgesic effect of morphine. The results were calculated as a percentage of maximal possible effect (%MPE) using the following formula: (drug response time – basal response time)/(10 seconds – basal response time) × 100.
Collection of cerebrospinal fluid
To confirm that morphine induces HMGB1 releasein vivo, we collected cerebrospinal fluid (CSF) from the experimental animals. Initially, we tried to collect CSF from mice, but found that it was difficult to do, and only 2–3 µL of CSF could be collected from each mouse. In contrast, it was relatively simple to collect CSF from rats; therefore, we collected CSF from a rat model of morphine-induced hyperalgesia. The rats were anesthetized with pentobarbital sodium (50 mg/kg, intraperitoneal injection; Sigma-Aldrich, St. Louis, MO, USA). The CSF (~80 µL) was carefully collected from the cisterna magna as described previously (Gholampour et al., 2022). After a short centrifugation step (5 minutes at 5000 ×g, 4°C), the samples were dissolved in 2× sodium dodecyl sulfate loading buffer, boiled, and analyzed by sodium dodecyl sulfatepolyacrylamide gel electrophoresis followed by western blotting. A schematic of these animal experiments is shown in Additional Figure 1.
Cell cultures
Human neuroblastoma SH-SY5Y cells (EK-Bioscience, Shanghai, China, Cat# CC-Y1459, RRID:CVCL_0019), identified by short tandem repeat analysis, were maintained in a humidified 5% CO2atmosphere at 37°C in Dulbecco’s modified Eagle medium/F-12 (Gibco, Grand Isl and, NY, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco), 80 U/mL penicillin, and 0.08 mg/mL streptomycin. For further experiments, SH-SY5Y cells (1 × 106cells per well) were plated in 6-well plates overnight and then treated the following morning with morphine (200 µM, Shenyang First Pharmaceutical Factory, Northeast Pharmaceutical Group Company) with or without zinc protoporphyrin IX (2 µM, Sigma-Aldrich) or Compound C (10 µM) for 12 hours. After that, the cell extracts and precipitated supernatants were collected.
BV-2 cells (EK-Bioscience, Shanghai, China, Cat# CC-Y2022, RRID: CVCL_5I31) are a microglial cell line derived from C57/BL6 mice and identified by short tandem repeat analysis. The cells were maintained in a humidified 5% CO2atmosphere at 37°C in Dulbecco’s modified Eagle medium (KenGEN BioTECH, Nanjing, Jiangsu, China) supplemented with 10% (v/v) fetal bovine serum, 80 U/mL penicillin, and 0.08 mg/mL streptomycin.
To test the level of HMGB1-mediated NF-κB p65 phosphorylation in the presences or absence of a TLR4 antagonist, BV-2 cells were plated in 6-well plates overnight and then treated with 25 nM recombinant HMGB1 protein (Novus Biologicals, Littleton, CO, USA) or normal IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The cells were pretreated with TLR4 antagonist (TAK242, 10 µM; MedChemExpress) for 15 minutes before treatment with recombinant HMGB1. To confirm the role of HMGB1 in triggering an inflammatory response, conditioned medium collected from SHSY5Y cells after morphine treatment (200 µM, 12 hours) was used to activate BV-2 cells in the presence of an anti-HMGB1 neutralizing antibody (2 µg/µL, Novus Biologicals) or normal IgG (2 µg/µL, Novus Biologicals). Then, the cell extracts and precipitated supernatants were analyzed by immunoblot assay. A schematic of these animal experiments is shown in Additional Figure 2.
Western blot
Under deep anesthesia induced by intraperitoneal injection with pentobarbital sodium (50 mg/kg), L1–L6 spinal cord tissue segments were collected from mice. Samples (cells or spinal cord tissue) were washed with ice-cold phosphate buffer saline before being lysed in radioimmunoprecipitation assay lysis buffer (Beyotime Biotechnology, Shanghai, China). CSF from rats was directly lysed in radio immunoprecipitation assay lysis buffer. After the protein concentrations were measured by bicinchoninic acid protein assay (Cortés-Ríos et al., 2020), the samples were heated for 5 minutes at 100°C. Then, the protein samples were loaded onto 8–15% sodium dodecyl sulfatepolyacrylamide gels and subjected to electrophoresis. Next, the proteins were electrophoretically transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 10% low-fat dry powdered milk or with 5% bovine serum albumin and 5% low-fat dry powdered milk (for phosphorylated protein) in Tris-HCl, NaCl, and Tween-20 for 2 hours at 20–25°C, and then probed with primary antibodies at 4°C for overnight. The primary antibodies utilized included β-actin (1:5000, mouse, Sigma-Aldrich, Cat# A5441, RRID: AB_476744), adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK; 1:1000, rabbit, Cell Signaling Technology, Danvers, IL, USA, Cat# 2532S, RRID: AB_330331), phospho (p)-AMPK (Thr172) (1:1000, rabbit, Cell Signaling Technology, Cat# 2535S, RRID: AB_331250), heme oxygenase-1 (HO-1; 1:1000, rabbit, Affinity Biosciences, Cincinnati, OH, USA, Cat# AF5393, RRID: AB_2837878), nuclear factor kappa-B p65 (NF-κB p65; 1:1000, rabbit, Cell Signaling Technology, Cat# 8242S, RRID: AB_10859369), p-NF-κB p65 (Ser536) (1:1000, rabbit, Cell Signaling Technology, Cat# 3033S, RRID: AB_331284), HMGB1 (1:1000, rabbit, Abcam, Cambridge, MA, USA, Cat# ab18256, RRID: AB_444360), transferrin (1:1000, rabbit, Abcam, Cat# ab84036, RRID: AB_10673794), and IL-1β (1:300, goat, R&D Systems, Minneapolis, MN, USA, Cat# AF-401-NA, RRID: AB_416684). β-Actin was used as an internal control, whereas all of the other target proteins listed above were used to investigate the mechanism of morphine tolerance. Finally, horseradish peroxidase–coupled secondary antibodies were utilized to detect the corresponding primary antibodies at 20–25°C for 2 hours. The following secondary antibodies were used: horseradish peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H+L) (1:5000, Sigma-Aldrich, Cat#AP307P, RRID:AB_11212848), horseradish peroxidase-conjugated AffiniPure goat anti-mouse IgG(H+L) (1:5000, Sigma-Aldrich, Cat# AP308P, RRID: AB_11215796), and horseradish peroxidase-conjugated AffiniPure donkey anti-goat IgG(H+L) (1:5000, Sigma-Aldrich, Cat# AP180P, RRID: AB_92573). The bands were then developed by enhanced chemiluminescence (PerkinElmer, Waltham, MA, USA). Data were analyzed using the Molecular Imager chemidocxrs system (Bio-Rad, Hercules, CA, USA) and the associated ImageJ software (version 1.51, National Institutes of Health, Bethesda, MD, USA; Schneider et al., 2012).
Immunofluorescence staining
Immunofluorescence staining was performed to investigate the localization and expression of HMGB1, ionized calcium-binding adaptor molecule 1 (Iba1), glial fibrillary acidic protein (GFAP), and NeuN. For fluorescence immunohistochemistry, mice were anesthetized and transcardially perfused with 4% cold paraformaldehyde on day 7. Lumbar spinal cords (L4–L5) were harvested, post-fixed for 4 hours at 4°C in 4% paraformaldehyde, and then cryoprotected sequentially in 10%, 20%, and 30% sucrose overnight for 3 days. Frozen sections (15 µm) were cut on a cryostat and air-dried on microscope slides for 30 minutes at room temperature. For dual antibody immunofluorescence, the tissue sections were incubated with primary antibodies against HMGB1 (1:200, rabbit, Abcam, Cat# ab18256, RRID:AB_444360), Iba1 (goat, 1:200, Abcam, Cat# ab5076, RRID: AB_2224402), NeuN (1:200, mouse, Millipore, Livingston, Scotland, UK, Cat# MAB377, RRID: AB_2298772), and GFAP (goat, 1:200, Millipore, Cat# MAB360, RRID: AB_11212597) in 10% normal donkey serum and 0.01% Triton-X-100 overnight at 4°C. For c-fos and calcitonin gene related peptide (CGRP) fluorescence immunohistochemistry, the tissue sections were incubated with primary antibodies against c-fos (1:300, rabbit, Cell Signaling Technology, Cat# 2250S, RRID: AB_2247211) and CRGP (1:300, rabbit, Cell Signaling Technology, Cat#14959, RRID: AB_2798662) in 10% normal donkey serum and 0.01% Triton-X 100 overnight at 4°C. The appropriate fluorescent secondary antibody was used for each primary antibody at 20–25°C for 2 hours. The following secondary antibodies were used: Alexa Fluor 488 AffiniPure donkey anti-rabbit IgG (H+L), Cy3 AffiniPure donkey anti-mouse IgG (H+L), and Cy3 AffiniPure donkey anti-goat IgG (H+L) (all 1:300, Jackson Immunoresearch Laboratories, West Grove, PA, USA, Cat# 711-545-152/715-165-150/705-165-003, RRID: AB_2313584/AB_2340813/AB_2340411). Confocal microscopy of immunofluorescence in the dorsal horn was performed using a confocal microscopy (Zeiss LSM710, Oberkochen, Germany).
RNA interference
HO-1 small interfering RNA (siRNA) (Cat# EHU051241, Sigma), AMPK siRNA (AMPKα1, Cat# EHU074041; AMPKα2, Cat# EHU042081, Sigma), and control siRNA (Cat# EHUEGFP, Sigma) were purchased from Sigma-Aldrich. Control siRNA (Cat# SIC001, Sigma) was used as a negative control. For siRNA transfection, SH-SY5Y cells were cultured in 6-well plates with antibiotic-free medium the day before transfection. The transfection was conducted when cells reached 50–70% confluency using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and serum-free medium according to the manufacturer’s instructions. SH-SY5Y cells were transfected with 100 pmol siRNA. After 5 hours, the Dulbecco’s modified Eagle’s medium was replaced with culture medium containing 10% fetal bovine serum, and the cells were then incubated at 37°C.
3-(4,5)-Dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumromide assay
SH-SY5Y cells were seeded in 96-well culture plates at a density of 5 × 103/well and incubated for 24 hours. Then, the cells were treated with various concentrations of morphine for another 12 hours. Cell viability was assessed using a 3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumromide Cell Cytotoxicity Assay Kit (Cat# C0009S, Beyotime Biotechnology) according to the manufacturer’s recommendations. The cell viability was calculated based on the ratio of optical density to that of the control group.
Statistical analysis
Sample sizes were determined based on previous studies (Cai et al., 2014; Qu et al., 2019). No animals were excluded from the analysis. The raters were blinded to the group assignments. GraphPad Prism 7.0a software (GraphPad Software, San Diego, CA, USA, www.graphpad.com) was used to conduct all the statistical analyses. Differences between two groups were evaluated by one-samplet-test. Data from more than two groups were evaluated by oneway analysis of variance or two-way analysis of variance accompanied by Tukey’s multiple-comparison test. Results are represented as mean ± standard error of the mean (SEM) of the independent experiments. The significance was based on a criterion ofP< 0.05.
Results
Morphine induces HMGB1 release in vivo and in vitro
Rats were intrathecally injected with morphine (10 µg/10 µL) once daily for 7 consecutive days to establish the morphine tolerance model, after which CSF was collected to investigate whether morphine induced HMGB1 efflux into the extracellular environment. The western blot results showed that morphine caused marked release of HMGB1 into CSF (Figure 1A). To further investigate the cellular mechanism underlying morphine-induced HMGB1 release, we analyzed HMGB1 distribution by confocal microscopy. In the control mice, HMGB1 was mainly localized in neurons (NeuN-positive cells), not in astrocytes (GFAP-positive cells) or microglia (Iba-positive cells). In addition, HMGB1 localized to cell nuclei, reflecting HMGB1’s role as a nonhistone nuclear protein. However, treatment with morphine resulted in a decrease in double-positive staining for HMGB1 and NeuN in the spinal cord of mice. Furthermore, the subcellular location of HMGB1 changed, strongly implying that morphine induced the release of HMGB1 from the nucleus into the extracellular CSF (Figure 1B). Next, the human neuroblastoma cell line SH-SY5Y, which expresses both mu- and delta-opioid receptors (at an approximate ratio of 4.5:1) (Yu et al., 1990) was utilized to providein vitroconfirmation of morphine-induced HMGB1 release. SH-SY5Y cells were incubated with different concentrations (50, 100, 200 µM) of morphine for 12 hours, and then the supernatants were analyzed by western blot. 3-(4,5)-Dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumromide assay confirmed that morphine did not affect cell proliferation at the different concentrations used (50, 100, and 200 µM) (Additional Figure 3). Western blotting showed that treatment with morphine induced HMGB1 excretion into the extracellular environment in a concentration-dependent manner (Figure 1C). Moreover, the immunofluorescence results showed that morphine caused HMGB1 to migrate from the nucleus to the cytoplasm (Figure 1D).
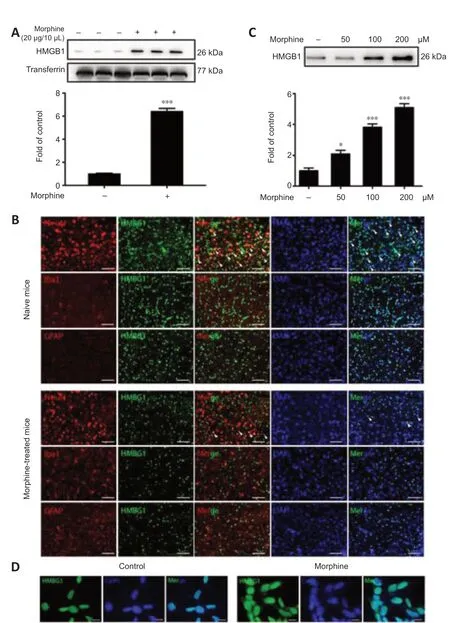
Figure 1|Morphine induces HMGB1 release.
Glycyrrhizin attenuates chronic morphine tolerance
Given our observations indicating that morphine induced HMGB1 release, next we examined whether the released HMGB1 was important for morphine tolerance. As a selective HMGB1 inhibitor (Li et al., 2019), glycyrrhizin was utilized to evaluate the effects of the released HMGB1 on morphine tolerance. Glycyrrhizin did not enhance the acute analgesic effect of morphine (Figure 2A), and the behavioral test results revealed that glycyrrhizin suppressed chronic morphine tolerance in a dose-dependent manner (Figure 2B). On day 7, the MPE at 30 minutes decreased to 14.5% in chronic morphine-treated mice, whereas mice that received both glycyrrhizin (25, 50, and 100 mg/kg) and morphine displayed MPEs of 26.3%, 35.9%, and 54.7%, respectively (Figure 2B). Moreover, high levels of c-fos (a marker of nociceptive neuron activation (Gao and Ji, 2009)) and CGRP (an indicator of pain (Benemei et al., 2009)) expression were observed in the dorsal horn of the spinal cord in mice with morphine tolerance, and this effect was inhibited by treatment with 50 or 100 mg/kg glycyrrhizin (Figure 2C).
Extracellular HMGB1 triggers an inflammatory response that is dependent on TLR4 expression in microglia
Next, we explored the role of the released HMGB1 in neuroinflammation. Microglia, which account for 10–15% of all cells in the central nervous system, are the resident immune cells in the central nervous system. When exposed to a variety of damaging stimuli, such as ischemia/hypoxia, trauma, and infection, microglia are rapidly activated and release a large number of proinflammatory cytokines, including TNF-α, IL-1β, and IL-6 (Walter et al., 2007; Fernandez-Lizarbe et al., 2013; Deora et al., 2020). It has been reported that microglial TLR4 activated by DAMPs potentiates neuroinflammation through inflammasome-induced IL-1β expression. Cai et al. (2016) reported that morphine induces NLRP3 inflammasome activation and IL-1β maturation. We found that repeated morphine administration led to phosphorylation of NF-κB p65 and upregulation of the proinflammatory cytokine IL-1β in the spinal cord (Figure 3A and B). Glycyrrhizin attenuated these effects (Figure 3A and B). Moreover, immunofluorescence analysis showed that repeated morphine treatment led to an increase in Iba-1 expression, and that glycyrrhizin inhibited microgligal activation (Figure 3C). In order to confirm the involvement of microglia in morphine-induced NF-κB p65 phosphorylation and IL-1β upregulation in the spinal cord, we pretreated mice with PLX3397 to deplete microglia. Mice were treated with PLX3397 (40 mg/kg) for a total of 10 days starting 3 days before the first injection of morphine. The results showed that PLX3397 significantly depleted microglia in mice (Additional Figure 4A) and suppressed chronic morphine tolerance (Additional Figure 4B). Furthermore, western blot analysis showed that PLX3397 decreased NFκB p65 phosphorylation and IL-1β expression in the spinal cord (Additional Figure 4C). These findings demonstrate that microglial activation is necessary for inducing morphine tolerance.
We next verified the effect of extracellular HMGB1 on the inflammatory response using the immortalized murine microglial cell line BV-2 (Blasi et al., 1990; Wang et al., 2012). We found that treatment with recombinant HMGB1 (25 nM) significantly upregulated NF-κB p65 phosphorylation in BV-2 cells, whereas treatment with a TLR4 antagonist (TAK242, 10 µM) inhibited this effect (Figure 3D). These findings show that microglial activation triggered by HMGB1-TLR4 signaling is necessary for morphine tolerance. Finally, we collected conditioned medium from SH-SY5Y cells treated with morphine (200 µM, 12 hours) and used it to activate BV-2 cells to confirm the role of HMGB1 in triggering an inflammatory response. Anti-HMGB1 neutralizing antibody (2 µg/µL) decreased the NF-κB p65 phosphorylation and upregulation of IL-1β expression induced by the conditioned medium (Figure 3E and F). Normal IgG (2 µg/µL) did not have any significant effect (Figure 3E and F).
Morphine-induced HMGB1 release is dependent on the AMPK/HO-1 pathway
Next, we asked how morphine induces HMGB1 release from neurons. Our data showed that morphine increases HO-1 expression in SH-SY5Y cells (Figure 4A). Pretreating the cells with zinc protoporphyrin IX (HO-1 inhibitor, 2 µM) for 12 hours before morphine (200 µM) administration suppressed the morphine-induced release of HMGB1 (Figure 4B). Furthermore, transfection with HO-1 siRNA downregulated HO-1 expression and inhibited the morphineinduced release of HMGB1 from SH-SY5Y cells (Figure 4C and D).
Recent studies indicated that HO-1 expression is regulated by several factors, such as silent information regulator 1 (SIRT-1), AMPK, and nuclear erythroid 2-related factor 2 (Nrf2) (Senthil et al., 2016; Park et al., 2018). AMPK, which is a key regulator of energy homeostasis, plays a fundamental role in chronic pain occurrence, development, and maintenance (Gui et al., 2018). Hence, we asked whether HO-1-HMGB1 activation was downstream of AMPK. Firstly, we found that AMPK phosphorylation increased after morphine administration in a concentration-dependent manner in SH-SY5Y cells, and that Compound C significantly decreased HO-1 expression and induced HMGB1 release in SHSY5Y cells (Figure 5A–C). Furthermore, transfection with AMPK siRNA resulted in decreased AMPK phosphorylation and HO-1 expression, and induced HMGB-1 release from SH-SY5Y cells (Figure 5D–F).
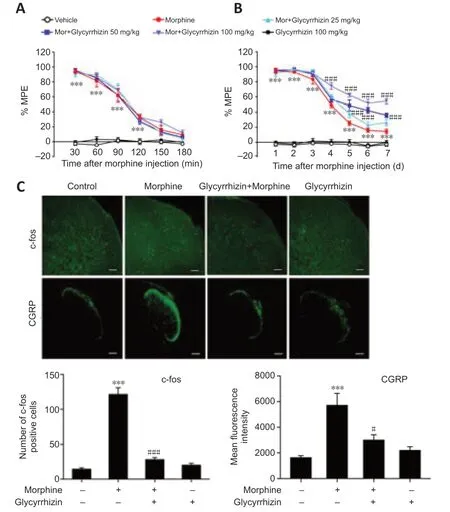
Figure 2|Glycyrrhizin attenuates chronic morphine tolerance.
To confirm these results, we administered repeated intrathecal injections of Compound C to evaluate the effect of AMPK on chronic morphine tolerancein vivo. Compound C not only dramatically potentiated acute morphine analgesia but also attenuated chronic morphine tolerancein vivo(Figure 6A and B). On day 7, the MPE at 30 minutes decreased to 14.39% in mice with chronic morphine tolerance, while co-administration of Compound C (1, 3, and 10 µg/10 µL) and morphine resulted in MPEs of 28.79%, 51.52%, and 70.05% respectively (Figure 6B). Furthermore, western blot analysis showed that Compound C decreased HMGB1 release into the CSF (Figure 6C) and suppressed p-AMPK and HO-1 expression levels. Furthermore, Compound C inhibited morphine-induced NF-κB p65 phosphorylation in the spinal cord (Figure 6D–F). Taken together, these results show that morphine induces HMGB1 release via the AMPK/HO-1 pathway and that suppression of this pathway can effectively improve chronic morphine tolerance.
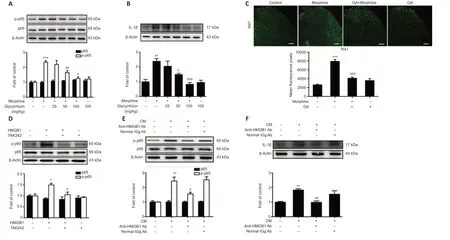
Figure 3|Extracellular HMGB1 triggers an inflammatory response that is dependent on TLR4 expression in microglia.
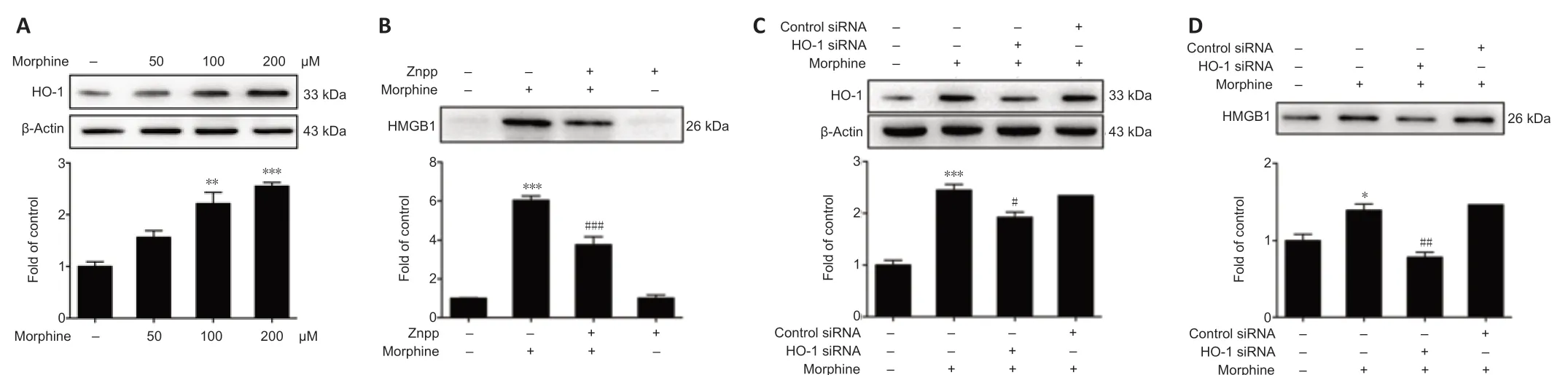
Figure 4|Morphine-induced HMGB1 release requires HO-1.
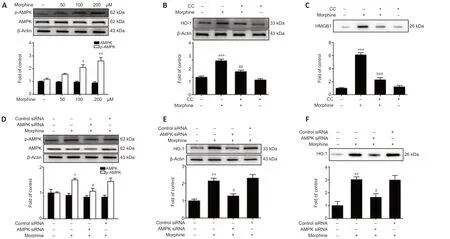
Figure 5|Morphine-induced AMPK phosphorylation increases HMGB1 release by upregulating HO-1.

Figure 6|Compound C potentiates acute morphine analgesia and suppresses chronic morphine tolerance.
Discussion
The mechanisms underlying morphine tolerance are not fully understood. Many mechanisms are involved in morphine tolerance, including desensitization or internalization of the opioid receptor, elevation of cAMP levels, and downregulation of spinal glutamate transporters (Christie, 2008). In particular, there is evidence that chronic morphine treatment induces sterile neuroinflammation in the spinal cord. HMGB1 is an important mediator of neuroinflammation in many pathophysiological conditions (Qian et al., 2020). For example, massive release of HMGB1 into the extracellular space after ischemic insult induces neuroinflammation in the postischemic brain (Kim et al., 2006). The HMGB1-macrophage antigen complex 1-nicotinamide adenine dinucleotide phosphate hydrogen pathway links neuroinflammation to progressive dopaminergic neurodegeneration in Parkinson’s disease (Gao et al., 2011). In addition, persistent HMGB1 release leads to tactile hyperalgesia in a rodent model of neuropathic pain (Feldman et al., 2012). Anti-HMGB1 monoclonal antibody has been shown to be effective for the treatment of a wide range of central nervous system diseases in animal models, including stroke, traumatic brain injury, epilepsy, and Alzheimer’s disease (Nishibori et al., 2019). Recently, Qian et al. (2020) discovered that morphine-mediated upregulation of HMGB1 in the spinal cord contributed to analgesic tolerance and hyperalgesia. In our study we found that morphine induces HMGB1 efflux into the extracellular environment via the AMPK-HO-1 axis. Furthermore, we showed that the released HMGB1 triggers TLR4 signaling, which consequently induces upregulation of NF-κB p65 phosphorylation and proinflammatory cytokine expression. Our immunoblot data demonstrate that HMGB1 is released into the CSF in response to treatment with morphine. Hence, we concluded that morphine-induced HMGB1 release most likely plays a critical role in linking neurons with microglia, especially during neuroinflammation. HMGB1 exerts its functions by binding to different receptors, including TLR2, TLR4, TLR5, TLR9, and receptors for advanced glycation end products (Andersson and Tracey, 2011). The HMGB1-TLR4 binding interaction is a strong trigger for pro-inflammatory cytokine production. Morphine administration has been reported to induce NLRP3 inflammasome activation, and subsequently IL-1β maturation (Cai et al., 2016). Our study provides direct evidence that morphine induces HMGB1 release from neurons, and that the released HMGB1 triggers neuroinflammation in a TLR4-dependent manner, thereby leading to morphine tolerance.
Next, we investigated the mechanism of morphine-induced HMGB1 release from neurons. It has been shown that HO-1 decreases HMGB1 expression during the inflammatory response. HO-1, a stress-responsive protein, serves a vital metabolic function as the rate-limiting step in the degradation of heme (Tenhunen et al., 1968). It may function as a pleiotropic regulator during inflammation. Increased HO-1 expression is normally associated with an inflammatory response or with oxidative stress. HO-1 can be upregulated by several factors, such as hypoxia, hyperoxia, heat shock, cytokines, hydrogen peroxide, ultraviolet irradiation, and its substrate heme (Taketani et al., 1989; Patel et al., 2003). Although many preclinical studies have suggested that HO-1 plays an anti-inflammatory role in tissue injury, recent studies also support a pro-pathogenic effect for HO-1 in the propagation of chronic inflammation. HO-1 is regarded as one of the strongest positive predictors of metabolic disease in humans (Ryter and Choi, 2016). Huang et al. (2013) reported that HO-1 overexpression in adipocytes did not protect against highfat diet-induced obesity or the development of insulin resistance in mice. In addition, conditional HO-1 deletion in hepatocytes and macrophages conferred protection against diet-induced insulin resistance and inflammation, dramatically reducing the development of secondary diseases such as steatosis and liver toxicity (Jais et al., 2014). Heterozygous Hmox1 knockout mice were also shown to be protected against high-fat diet-induced insulin resistance because of reduced macrophage migration (Huang et al., 2012). Therefore, the complex role of the HO-1-HMGB1 axis in the development of morphine tolerance remains incompletely understood. Our results indicate that morphine induces HMGB1 release via HO-1 activation and that zinc protoporphyrin IX and HO-1 siRNA significantly decrease morphine-induced HMGB1 release.
AMPK is considered a novel target for the treatment of pain (Asiedu et al., 2016). Zhang et al. (2020a) demonstrated that morphine activates the AMPK pathway, induces epithelial-mesenchymal transition, and increases oxidative stress in esophageal carcinoma cells by upregulating Snail and Slug levels. In addition, morphine inhibits PTEN-induced putative kinase 1/Parkin-associated mitophagy and increases AMPK phosphorylation (Kong et al., 2019) in spinal cord neurons. HO-1 expression is controlled by AMPK (Shen et al., 2019; Liu et al., 2021), and AMPK activation increases HO-1 expression. We found that morphine increases AMPK phosphorylation. Thus, our findings show that morphine-induced HMGB1 release is most likely dependent on the AMPKHO-1 axis. However, our study had some limitations. The mechanism underlying AMPKmediated regulation of HO-1 expression needs to be investigated in the future. Nrf2, a downstream gene of AMPK, has been reported to play an important role in regulating HO-1 expression. AMPK phosphorylates Nrf2 at the Ser550 residue, which promotes Nrf2 accumulation in the nucleus, where, as a regulator of the antioxidant system, it upregulates HO-1 expression (Joo et al., 2016; Zhang et al., 2020b). At present, it is unclear whether inhibiting Nrf2 could alleviate morphine tolerance. In our study, we showed that inhibiting the AMPK-HO-1-HMGB1 axis alleviates morphine tolerance via inhibiting neuroinflammation. However, some studies have shown that AMPK/Nrf2/HO-1 signaling has a protective, anti-inflammatory effect in various diseases. Lei et al. (2020) reported that, in lipopolysaccharide-induced endotoxemic mice, activating AMPK/Nrf2/HO-1 signaling in macrophages attenuated inflammation. In addition, Park et al. (2018) reported that upregulating the p-AMPK/Nrf2/HO-1 signaling pathway in astrocytes had a neuroprotective effect in mice with MPTP-induced Parkinson’s disease. Therefore, the AMPK/Nrf2/HO-1 axis may play different roles in different diseases, and confirming the safe dose range for suppressing morphine tolerance needs to be explored in more detail. Another limitation is the limitation of experiment design. We only used male rodents to investigate the mechanism of morphine tolerance. However, there are sex differences in morphine tolerance, which merits further study to determine the underlying mechanisms and clinical implications.
In summary, our study illustrated that morphine-induced release of HMGB1 into extracellular space is critical for morphine tolerance. HMGB1 release from neurons is mediated by the AMPK-HO-1 pathway, and the released HMGB1 mediates morphine-induced activation of TLR4 in microglia. The extracellular HMGB1 increases NF-κB p65 phosphorylation via TLR4 and promotes expression of the proinflammatory cytokine IL-1β. Therefore, inhibiting HMGB1 and/or the AMPK-HO-1 axis may be a viable approach for treating patients with morphine tolerance.
Author contributions:Study design: CYJ, WTL; experimental implementation: TTL, LS, LW, WF, JCL, XDS, YBP; data analysis: TTL, CJX, LH, XFW, YH; manuscript draft: CYJ, TTL, LS. All of the authors reviewed and approved the final version of manuscript.
Conflicts of interest:The authors declare that they have no competing interests.
Author statement:This paper has been posted as a preprint on Research Square with doi: https://doi.org/10.21203/rs.3.rs-126767/v1, which is available from: https://assets.researchsquare.com/files/rs-126767/v1/b3725c30-12df-4762-8883-5df844331b30.pdf?c=1631867487.
Data availability statement:All relevant data are within the paper and its Additional files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional Figure 1:Schematic diagram of the in vivo experiments.
Additional Figure 2:Schematic diagram of the in vitro experiments.
Additional Figure 3:The different concentrations of morphine that did not affect cell proliferation.
Additional Figure 4:PLX3397 alleviates chronic morphine tolerance.

- 中国神经再生研究(英文版)的其它文章
- Corrigendum
- Extracellular vesicles as a potential therapeutic for agerelated macular degeneration
- Survival of rat sciatic nerve segments preserved in storage solutions ex vivo assessed by novel electrophysiological and morphological criteria
- Fasting produces antidepressant-like effects via activating mammalian target of rapamycin complex 1 signaling pathway in ovariectomized mice
- Transcriptional regulatory network during axonal regeneration of dorsal root ganglion neurons: laser-capture microdissection and deep sequencing
- 5-Hydroxytryptamine: a potential therapeutic target in amyotrophic lateral sclerosis