
miR-181b promotes angiogenesis and neurological function recovery after ischemic stroke
2023-02-13 12:41LiXiaXueLinYuanShuHongMeiWangKaiLiLuLiGangHuangJingYanXiangZhiGengYuWuZhaoHaoChen
中国神经再生研究(英文版) 2023年9期
Li-Xia Xue , Lin-Yuan Shu , Hong-Mei Wang Kai-Li Lu Li-Gang Huang Jing-Yan Xiang Zhi Geng Yu-Wu Zhao Hao Chen
Abstract Promotion of new blood vessel formation is a new strategy for treating ischemic stroke. Non-coding miRNAs have been recently considered potential therapeutic targets for ischemic stroke. miR-181b has been shown to promote angiogenesis in hypoxia and traumatic brain injury model, while its effect on ischemic stroke remains elusive. In this study, we found that overexpression of miR-181b in brain microvascular endothelial cells subjected to oxygen-glucose deprivation in vitro restored cell proliferation and enhanced angiogenesis. In rat models of focal cerebral ischemia, overexpression of miR-181b reduced infarction volume, promoted angiogenesis in ischemic penumbra, and improved neurological function. We further investigated the molecular mechanism by which miR-181b participates in angiogenesis after ischemic stroke and found that miR-181b directly bound to the 3′-UTR of phosphatase and tensin homolog (PTEN) mRNA to induce PTEN downregulation, leading to activation of the protein kinase B (Akt) pathway, upregulated expression of vascular endothelial growth factors, down-regulated expression of endostatin, and promoted angiogenesis. Taken together, these results indicate that exogenous miR-181b exhibits neuroprotective effects on ischemic stroke through activating the PTEN/Akt signal pathway and promoting angiogenesis.
Key Words: Akt; angiogenesis; endostatin; ischemic stroke; middle cerebral artery occlusion; miR-181b; neurological function recovery; oxygen-glucose deprivation; PTEN; vascular endothelial growth factor
Introduction
Ischemic stroke (IS), which accounts for 87% of all strokes, is a leading cause of mortality and long-term disability worldwide (Yousufuddin and Young, 2019). Currently, thrombolytic therapy and endovascular mechanical clot removal are effective recanalization interventions for IS, but their clinical application is relatively limited because of the narrow therapeutic window and potential risk of hemorrhagic transformation (Kong et al., 2021). Neuroprotection, even though a major focus of IS therapy research, remains controversial due to the lack of promising evidences in clinical trials (Paul and Candelario-Jalil, 2021). Therefore, other effective treatments are urgently required.
After IS, angiogenesis is activated to modify the cerebral microvasculature and collateral circulation and is an effective way to restore the reduced cerebral perfusion, thereby promoting the recovery of neurological functions (Ergul et al., 2012). Some therapeutic drugs for IS, such as butylphthalide, have shown effects in increasing collateriogenesis after focal IS in animal experiments (Wei et al., 2021). Our previous clinical trial also demonstrated that butylphthalide is more effective for IS treatment than a neuroprotective peptide, benefiting from its dual activities of neuroprotection and angiogenesis (Xue et al., 2016). These findings suggest the therapeutic potential of angiogenesis-related treatments for IS (Maacha et al., 2020).
MicroRNAs (miRNAs) are abundantly expressed in the central nervous system and function as pivotal modulators of brain development and function (Chen et al., 2022; Zhang et al., 2022). miRNAs have also emerged as potential biomarkers or therapeutic targets for IS (Maacha et al., 2020). Several miRNAs, including miR-124, miR-126, miR-137, miR-145, miR-126, miR-223, and miR-493, were reported to play an important role in neurological functional recovery after IS through regulating apoptosis, neuroinflammation, neurogenesis, and angiogenesis in preclinical models of IS (Qu et al., 2019; Qian et al., 2020; Gugliandolo et al., 2021). Since the neuroprotective therapy involving miRNAs is promising, further studies are needed to uncover the miRNAs that may exert neuroprotective functions in IS and advance the clinical translation of miRNA-based therapy.
miR-181b, a hypoxia-regulated mRNA, was reported to enhance angiogenesis via hypoxia in a HIF-1α-independent manner (Zhang et al., 2020), which is crucial for angiogenesis in IS. Recent studies showed that exosomederived miR-181b promotes angiogenesis of brain microvascular endothelial cells (BMVECs) after hypoxiain vitro(Yang et al., 2018) and inhibits neuroinflammation after traumatic brain injuryin vivo(Wen et al., 2022). Whether miR-181b participates in the endothelial response to IS, and exerts neuroprotective effects post-IS remains poorly defined.
Phosphatase and tensin homolog (PTEN), a tumor suppressor, is an inhibitor of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway and a crucial mediator of angiogenesis, as it activates vascular endothelial growth factor (VEGF) expression in tumorigenesis and vascular malformation (Huang et al., 2021). Growing evidence suggests that PTEN mediates neuronal death, endothelial cell proliferation, and migration after ischemiain vitro(Huang et al., 2021); PTEN suppression has been reported to promote neuroprotective effects in animal models of focal IS (Guo et al., 2013). We previously demonstrated that PTEN inhibition enhances angiogenesis in vascular endothelial cells through the promotion of Akt phosphorylation and subsequent HIF-1α upregulation after exposure to oxygen-glucose deprivation (OGD), anin vitromimic of stroke conditions (Xue et al., 2018). Using TargetScan software, we identified PTEN as a target gene of miR-181b through its binding to sites in the 3′-untranslated region (3′-UTR) of PTEN. Moreover, miR-181b suppresses PTEN expression and increases Akt phosphorylation in hepatic stellate cells (Geng et al., 2020). However, whether miR-181b contributes to the regulation of post-stroke angiogenesis by interacting with PTEN/Akt signaling has not yet been elucidated.
In this study, we tested the hypothesis that miR-181b protects BMVECs and promotes angiogenesis from OGD-induced injury by regulating the PTEN/Akt pathway. We also determined the potential effect of miR-181b on post-stroke angiogenesis in ischemic brain tissuesin vivo.
Methods
Animals
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the Shanghai Sixth People’s Hospital (approval No. 2018-0051) on February 27, 2018 and were performed in accordance with the Guide for the Care and Use of Laboratory Animals (8thed; National Research Council, 2011). Estrogen has been proven to have a neuroprotective effect in animal models of global and focal ischemia (Merchenthaler et al., 2003), so only male rats were included in the present study. Six-week-old male Sprague-Dawley rats (weight 250–280 g) were purchased from SPF (Beijing) Biotechnology Co., Ltd. (Beijing, China, license No. SCXK (Jing) 2019-0010). All rats were kept in individual standard cages under a 12-hour light/dark cycle at temperature of 23 ± 2°C and humidity of 55 ± 5%, with full access to food and water. Seventy-eight rats were randomly assigned to six groups, with thirteen rats in each group: normal control (NC), middle cerebral artery occlusion (MCAO) model, MCAO + miR-181b-5p agomir-Ctrl (agomir-Ctrl), MCAO + miR-181b-5p agomir (agomir), MCAO + miR-181b-5p antagomir-Ctrl (antagomir-Ctrl), and MCAO + miR-181b-5p antagomir (antagomir).
MCAO model
The MCAO model was constructed in accordance with a previously described protocol (Bleilevens et al., 2013). All procedures were performed under aseptic conditions. After 3 days of acclimation, rats were anesthetized with 4% isoflurane (MilliporeSigma, Burlington, MA, USA, Cat# PHR2874) in 70% N2/30% O2using a mask. The left common carotid artery, external carotid artery, and internal carotid artery were isolated. The common and external carotid arteries were ligated, and the distal terminal of the internal carotid artery was closed with a vascular clamp. A nylon thread was inserted into the internal carotid artery and gently advanced to occlude the middle cerebral artery; the rats were disinfected and maintained at 37°C. After 2 hours of occlusion, the suture was carefully removed to achieve blood flow reperfusion for another 24 hours before administration. Rats that did not exhibit contralateral paralysis were excluded from the study.
Administration of agomir and antagomir
The micrONTMrno-miR-181b-5p agomir control, MicrONTMrno-miR-181b-5p agomir, micrOFFTMrno-miR-181b-5p antagomir control, and micrOFFTMrno-miR-181b-5p antagomir were designed and synthesized by RiboBio (Guangzhou, China); the sequences are listed in Table 1. At 24 hours after MCAO, rats in different groups were injected via the caudal vein with the respective oligonucleotides (200 nmol/rat for once injection). The rats were injected every 5 days four times over 3 weeks. The treatment design is illustrated in Figure 1A. Three rats from each group were sacrificed for immunoblotting, and the other ten were subjected to neurological deficiency evaluation. After neurological deficiency evaluation, rats were sacrificed for infarction volume assay (five rats) or immunofluorescence (five rats).
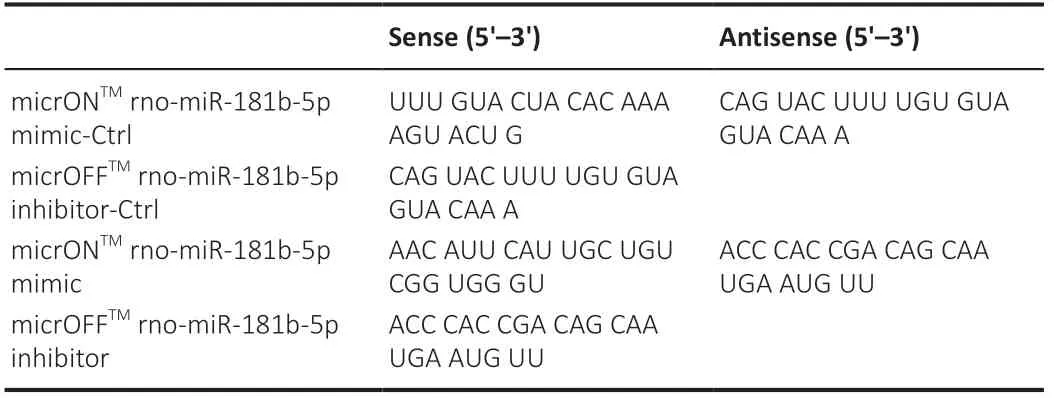
Table 1 |RNA sequences used in transfection
Evaluation of neurological deficiency
Zea Longa scores (0, no neurological deficit; 1, failure to extend the right forelimb; 2, circling to the right; 3, falling to the right; and 4, unable to walk spontaneously) were used for neurological deficit assessment on days 1 (before treatment), 7, 14, and 21 after MCAO, following Longa’s criteria (Longa et al., 1989). The rats were prevented from eating but not from drinking after the last administration of the oligonucleotides for 12 hours.
Infarction volume assay
Infarction volume assay was performed by 2,3,5-triphenyltetrazolium chloride staining. Following isoflurane anaesthetization, five rats from each group were sacrificed and their brains were collected. The brain samples were sectioned coronally at 2–3 mm intervals and stained with 2,3,5-triphenyltetrazolium chloride (Solarbio, Beijing, China, Cat# G3005) for 35 minutes in the dark. The sections were then fixed with 4% paraformaldehyde for 30 minutes. The cerebral infarction volume was analyzed using the IPP image analysis system (Media Cybernetics, Rockville, MD, USA) after absorbing moisture on the surface of the tissue, and the infarct volume ratio was calculated by dividing the infarct volume by the total volume of the slices.
Immunofluorescence
After neurological deficiency evaluation, rats were anesthetized by isoflurane and perfused with 50 mL phosphate-buffered saline (PBS) and 50 mL formalin intracardially. The brain was then taken out carefully from the skull, embedded in paraffin, and made into successive coronal sections of 20 µm by a microtome (Leica, Wetzlar, Germany). After embedding onto slides, brain slices were rinsed three times with PBS and then blocked with 1% bovine serum albumin for 30 minutes. The specimens were incubated with rabbit anti-CD31 antibody (1:50, Absin Bioscience Inc., Shanghai, China, Cat# abs131189; RRID: AB_2923365) overnight at 4°C, followed by incubation with Alexa 594-labeled goat anti-rabbit IgG (1:500, Beyotime Biotechnology, Shanghai, China, Cat# A0208, RRID: AB_2892644) for 2 hours at 37°C. To visualize nuclei, staining with 4′,6-diamino-2-phenyl indole (BBI Life Sciences, Shanghai, China, Cat# E607303) was performed for 5 minutes. Images were obtained by a laser scanning confocal microscope (Leica). For each brain, three equispaced sections were selected from the ischemic penumbra; for each section, three random microscope fields were analyzed to calculate the mean immunofluorescence intensity, which indicates the level of angiogenesis.
Cell culture and construction of the in vitro OGD model
Human BMVECs were purchased from BeNa Culture Collection Co. Ltd. (Beijing, China, Cat# BNCC350771, RRID: CVCL_YJ35) and maintained in RPMI 1640 medium (L260, Shanghai BasalMedia Technologies Co., LTD., Shanghai, China) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, NM, USA, Cat# 10099141) and 1% penicillin/streptomycin (5000 U/mL, Gibco) at 37°C in a 5% CO2incubator. Cells were authenticated by short tandem repeat methodology.
Thein vitroOGC model was constructed as previously described (Xue et al., 2018). BMVECs were transferred to an airtight experimental hypoxia chamber flushed with a gas mixture of 94% N2, 1% O2, and 5% CO2and the culture medium was replaced with glucose-free medium (RPMI 1640 medium without fetal bovine serum) for 8 hours. After terminating OGD, BMVECs were transfected with miR-181b-5p mimic and inhibitor for further experiments under normal growth conditions. BMVECs not exposed to OGD served as controls.
Transfection of miR-181b-5p mimic and miR-181b-5p inhibitor
The rno-miR-181b-5p mimic and rno-miR-181b-5p inhibitor were designed and synthesized by RiboBio; the sequences are listed in Table 1. Transfection of the miR-181b-5p mimic (100 pmol) and inhibitor (100 pmol) was performed using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA, Cat# 11668019), following the manufacturer’s instructions. miR-181b-5p mimic-Ctrl and miR-181b-5p inhibitor-Ctrl were transfected as controls. The transfected cells were cultured for 48 hours and used in subsequent experiments.
Cell proliferation, cell apoptosis, cell migration and in vitro angiogenesis assays
Cell proliferation was assessed using a Cell Counting Kit-8 assay kit (Beyotime Biotechnology, Cat# C0038) following the manufacturer’s instructions. Briefly, OGD-induced BMVECs subjected to different treatments were collected and seeded into 96-well plates overnight at a density of 2000 cells/well. After a 24-hour incubation, 10 µL of Cell Counting Kit-8 solution was added to each well and the sample was incubated for another 1 hour at 37°C. Cell proliferation was calculated by measuring the absorbance at 450 nm using a microplate reader (Biotek, Winooski, VT, USA).
Cell apoptosis was determined by flow cytometry using the Annexin V-FITC/PI Apoptosis Detection Kit (Beyotime Biotechnology, Cat# C1062) following the manufacturer’s instructions. Briefly, after 24 hours of incubation, cells were harvested and re-suspended in 195 µL of binding buffer. Then, 5 µL of Annexin V-FITC and 10 µL of propidium iodide were added to the binding buffer. The mixture was then gently mixed and incubated for 10 minutes at approximately 20°C in the dark. The cells were then analyzed by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA). The apoptotic rate was defined as the proportion of cells in Q2. For cell migration assays, Transwell inserts (Corning Costar, Corning, NY, USA) were inserted in a 48-well culture plate. OGD-induced BMVECs were seeded into the top chamber membrane at 1 × 105cells in 100 µL serum-free medium. The bottom chambers were filled with normal medium. After 48 hours, the migrated cells on the lower surface of the membrane were fixed with methanol for 20 minutes and stained with crystal violet (Sangon Biotech, Shanghai, China, Cat# E607309-0100) for 10 minutes. Cells on the upper surface were gently removed using a swab. The membranes were observed under a phase-contrast microscope (Leica), and the migration rates were analyzed by counting the number of migrated cells.
Forin vitroangiogenesis assays, BMVECs were seeded in 24-well plates covered with Matrigel (BD Biosciences, Cat# 356231) at a density of 3 × 105cells per well. BMVECs were then cultured in oxygen deficit or normoxic condition for 3 hours. Tube length and lumen number were quantified using ImageJ software (version 1.52a; National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012).
Dual luciferase reporter assay
Starbase (https://starbase.sysu.edu.cn/index.php) and miRDB databases (http://mirdb.org/policy.html) were used to predict the potential binding site of miR-181b-5p in the 3′-UTR of the wild-type (WT) PTEN gene (Chen and Wang, 2020). The WT 3′-UTR of PTEN were cloned downstream of the luciferase coding sequence in the psiCHECKTM-2 vector (OBiO, Shanghai, China). We also generated a mutant (Mut) vector in the same procedure, except that the predicted binding sites in the PTEN 3′-UTR were mutated. One day before transfection, 293T cells (Shanghai Zhong Qiao Xin Zhou Biotechnology, Shanghai, China, Cat# ZQ0033, RRID: CVCL_0063) were plated in 96-well plates so that they achieved 70% confluence at the time of transfection. The 293T cell line was authenticated using short tandem repeat methodology. The luciferase reporter vector (containing either the WT or Mut PTEN 3′-UTR), miR-181b-5p mimic, and mimic control were co-transfected into 293T cells. After 48 hours, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA, Cat# E1910), following the manufacturer’s instructions. Plasmid maps of PTEN and psiCHECKTM-2 plasmid are shown in Additional Figure 1.
Western blot analysis
Rats were sacrificed 3 weeks after MCAO; brain samples around the ischemic penumbra were added to lysis buffer, followed by homogenization by ultrasonic crushing. For cultured cells, lysis buffer was added to the culture plate, and a cell scraper was used to aid lysis. Protein concentrations were quantified using a bicinchoninic acid assay (Sangon Biotech, Cat# C503021). Samples were adjusted to 2 mg/mL before sample loading. Western blotting was performed following standard protocols. Briefly, after sodium dodecyl sulfate-polyacrylamide gel electrophoresis, the samples were transferred to a polyvinylidene fluoride membrane. The membranes were blocked with 5% milk in phosphate-buffered saline for 1 hour and incubated with primary antibodies at 4°C overnight. The membranes were rinsed three times and then incubated with the corresponding secondary antibody for 1 hour at room temperature. After rinsing with PBST for 10 minutes, immunoreactivity was visualized using the SuperSignal West Pico Kit (Thermo Fisher Scientific, Cat# 34077). Protein bands were quantified using ImageJ and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The antibodies were as follows: rabbit anti-PTEN antibody (1:1000, Abcam, Cambridge, UK, Cat# ab267787, RRID: AB_2923364), mouse anti-Akt antibody (1:2000, Proteintech, Wuhan, China, Cat# 60203-2-Ig, RRID: AB_10912803), rabbit anti-phosphorylated-(T308)-Akt antibody (1:1000, Abcam, Cat# ab38449, RRID: AB_722678), mouse anti-VEGF antibody (1:1000, Proteintech, Cat# 66828-1-Ig, RRID: AB_2882171), rabbit anti-endostatin (ES) antibody (1:500, Proteintech, Cat# 18301-1-AP, RRID: AB_2081719), rabbit anti-cleaved caspase-3 antibody (1:1000, Cell Signaling, Danvers, MA, USA, Cat# 9661, RRID: AB_2341188), rabbit/mouse anti-cleaved caspase-8 antibody (1:1000, Cell Signaling, Cat# 9429, RRID: AB_2259431), rabbit anti-cleaved caspase-9 antibody (1:1000, Cell Signaling, Cat# 9507, RRID: AB_2228625), rabbit anti-Bax antibody (1:5000, Abcam, Cat# ab32503, RRID: AB_725631), rabbit anti-Bcl-2 antibody (1:1000, Abcam, Cat# ab32124, RRID: AB_725644), and mouse anti-GAPDH antibody (1:8000, Proteintech, Cat# 60004-1-1, RRID: AB_2107436). The secondary antibodies used were horseradish peroxidaselabeled goat anti-mouse IgG (H+L) (1:10,000, Beyotime Biotechnology, Cat# A0216, RRID: AB_2860575) and horseradish peroxidase-labeled goat-antirabbit IgG (H+L) (1:10,000, Beyotime Biotechnology, Cat# A0208, RRID: AB_2892644).
Quantitative polymerase chain reaction
OGD-induced BMVECs were lysed using TRIzol reagent (Thermo Fisher Scientific, Cat# 15596026). miRNAs were isolated using miRNA extraction kits (Vazyme, Nanjing, China, Cat# RC201) following the manufacturer’s instructions. miRNA was reverse-transcribed using miRNA reverse kits (Qiagen, Dusseldorf, Germany, Cat# 218161) and the qRT-PCR Starter Kit (R1008, RiboBio) was used for quantitative polymerase chain reaction (qPCR). The reaction conditions were an initial enzyme activation step of 10 minutes at 95°C, followed by 50 cycles of 15 seconds at 95°C and 60 seconds at 60°C. Gene expression was calculated as a percentage of GAPDH mRNA. The qPCR primers used were as follows: miR-181b-5p forward primer, 5′-AAC AUU CAU UGC UGU GGG UGG GU-3′; miR-181b-5p reverse primer, 5′-CCA CCG ACA GCA AUG AAU GUU UU-3′; U6 primer, 5′-TTC GTG AAG CGT TCC ATA TTT T-3′; GAPDH forward primer, 5′-TGG CCT CCA AGG AGT AAG AAA C-3′; and GAPDH reverse primer, 5′-GGC CTC TCT CTT GCT CTC AGT ATC-3′.
Statistical analysis
Data are expressed as means ± standard deviation (SD). All experiments were analyzed using one-way analysis of variance followed by least significant difference test, andP< 0.05 was considered significant. SPSS 24.0 (IBM, Armonk, NY, USA) and GraphPad Prism 8.0.2 (GraphPad Software, San Diego, CA, USA, www.graphpad.com) were used for statistical analysis and visualization, respectively.
Results
Exogenous miR-181b-5p agomir rescues neurological function, reduces infarction volume, and promotes angiogenesis after MCAO in rats
We first explored the effects of miR-181b-5p agomir on IS. Intracerebroventricular or intravenous administration of rats with miR-181b-5p mimic/inhibitor or agomir/antagomir was shown to lead to overexpression or reduction of miR-181b-5p in rat brain (Wang et al., 2019; Chen et al., 2020). Therefore, we administered exogenous miR-181b-5p agomir or antagomir through intravenous injection in a rat MCAO model and analyzed six groups: NC, MCAO model, agomir-Ctrl, agomir, antagomir-Ctrl, and antagomir groups (Figure 1A). Before treatment, the NC group exhibited no neurological deficits while the other five groups had the same Zea Longa scores (P> 0.05). On day 14 after MCAO, rats in the miR-181b-5p antagomir group had higher Zea Longa scores than those in the NC group (P< 0.001), while the miR-181b-5p agomir treatment group demonstrated significant mitigation of neurological damage compared with that of the miR-181b-5p antagomir group (P< 0.01; Figure 1B). This result indicated that miR-181b-5p treatment successfully rescued neurological function in stroke rats. Cerebral infarct volume evaluation was performed using TCC staining and the results showed that MCAO induced a significant brain infarction (P< 0.001, compared with NC group; Figure 1C and D). Administration of the miR-181b-5p agomir effectively reduced the infarct area ratio (P< 0.001, compared with agomir Ctrl group), while administration of the miR-181b-5p antagomir increased infarct area ratio (P< 0.001, compared with antagomir Ctrl group; Figure 1C and D).
We also measured microvessel density by staining for CD31, a microvessel marker. MCAO rats administered miR-181b-5p agomir showed a higher CD31-positive rate in the brain compared with controls while the miR-181b inhibition group showed a lower density compared with controls (Figure 2A and B). This suggests that miR-181b stimulates angiogenesis in the rat brain after MCAO and inhibition of miR-181b suppresses MCAO-induced angiogenesis. To further explore the possible mechanism, we detected the expressions of PTEN/Akt pathway and the factors of angiogenesis. After administration of miR-181b-5p agomir, p-Akt and VEGF expressions were increased but PTEN and ES expressions were decreased, suggesting that miR-181b-5p-induced PTEN inhibition promotes angiogenesis probably through activating the Akt pathway and regulating the expressions of VEGF and ES in MCAO rats (Figure 2C and D).
miR-181b rescues cell proliferation and promotes angiogenesis in OGDinduced BMVECs
miR-181b has been demonstrated to enhance angiogenesis in retinoblastoma cells under hypoxic conditions (Xu et al., 2015). To test whether miR-181b plays a similar role in IS, we performed OGD on BMVECsin vitro. We used BMVECs in experiments with high purity and nearly 100% CD31-positive rates (Additional Figure 2). To evaluate the role of miR-181b in cell proliferation, apoptosis, and migration in OGD-induced BMVECs, we examined cells after transfection with miR-181b-5p mimic or miR-181b-5p inhibitor. Overexpression of miR-181b boosted cell proliferation and migration compared with controls, whereas inhibition of miR-181b decreased proliferation and migration (Figure 3A–C). Flow cytometry results demonstrated that transfection of the miR-181b-5p mimic in OGDinduced BMVECs decreased cell apoptosis rates, while inhibition of miR-181b increased apoptosis (Figure 3D and E). We confirmed that OGD treatment damaged angiogenesis in BMVECs (Figure 3F). Transfection of the miR-181b-5p mimic promoted tube formation ability to rescue angiogenesis in OGDinduced BMVECs, while inhibition of miR-181b reduced angiogenesis (Figure 3F–H).
miR-181b suppresses the expression of PTEN via binding to the 3'-UTR of PTEN mRNA
To explore the possible mechanisms of miR-181b in mediating neuroprotection and angiogenesis, we used StarBase and miRDB to predict the target genes of miR-181b. From the bioinformatic prediction results, PTEN was selected as a candidate for miR-181b. PTEN is involved in many cellular processes, including cell survival, proliferation, migration, energy metabolism, and cellular architecture (Song et al., 2012). PTEN also regulates angiogenesis through the PI3K/Akt/VEGF signaling pathway (Ma et al., 2009). We confirmedthe efficacy of the miR-181b-5p mimic and inhibitor in BMVECs using qPCR (Figure 4A). PTEN was downregulated after transfection of the miR-181b-5p mimic in BMVECs, while inhibition of miR-181b-5p stimulated the expression of PTEN (Figure 4B and C). To examine whether miR-181b-5p regulates the expression of PTEN mRNA through direct binding, we predicted the potential binding sites in the 3′-UTR of PTEN mRNA using StarBase and miRDB (Figure 4D) and cloned the PTEN 3′-UTR region downstream of the luciferase gene in a luciferase reporter. We also generated a mutant reporter in which the predicted binding sites in the PTEN 3′-UTR were mutated. Transfection with miR-181b-5p mimic reduced luciferase expression of the vector containing the wild-type PTEN 3′-UTR; however, it had no effect on the mutant vector (Figure 4E). These results indicate that PTEN is a direct target gene of miR-181b and miR-181b regulates PTEN expression by binding to its 3′-UTR.
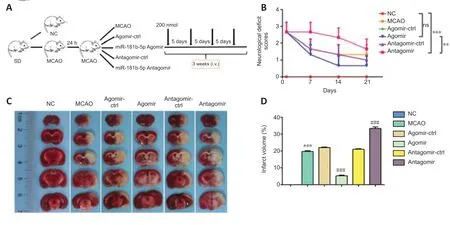
Figure 1|miR-181b-5p agomir promotes neurological function recovery and reduces infarct volume in MCAO rats.
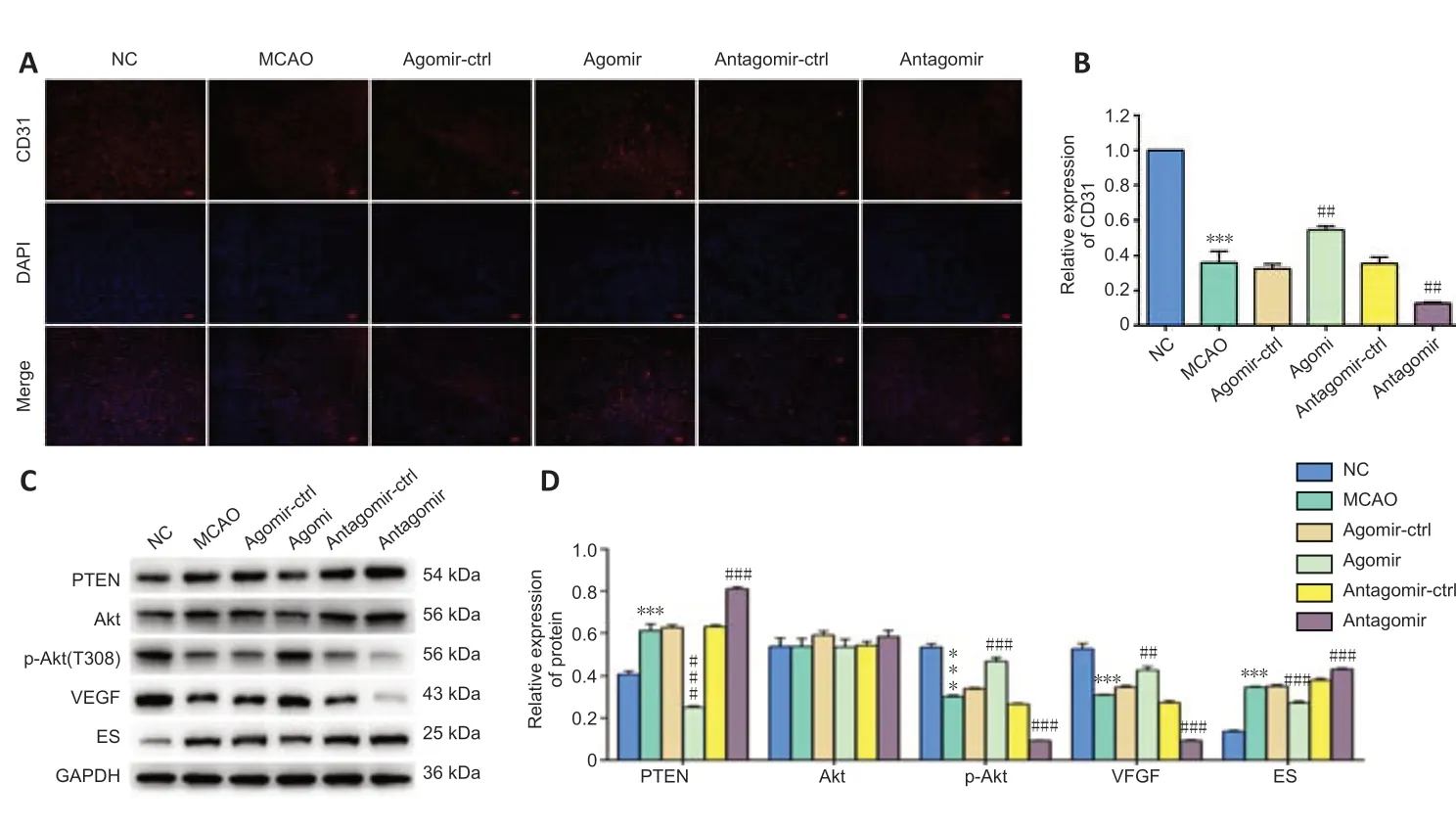
Figure 2|miR-181b-5p agomir promotes angiogenesis in rat brain after MCAO.
miR-181b activates the Akt signal pathway through inhibition of PTEN
PTEN inhibits angiogenesis by diminishing phosphorylation of Akt (Wen et al., 2001). Activation of the Akt pathway induces VEGF expression, a key mediator of angiogenesis during embryogenesis and tumorigenesis (Shiojima and Walsh, 2002; Carmeliet, 2005). Increased VEGF levels boost vascular permeability and angiogenesis, and ES, first identified as an angiogenesis inhibitor in 1997 (O’Reilly et al., 1997), potently inhibits endothelial proliferation and angiogenesis. ES and VEGF are both downstream effector molecules of angiogenesis. We found that OGD decreased p-Akt expression in BMVECs, indicating that inhibition of the Akt pathway might suppress angiogenesis (Figure 5A and B). After transfection with the miR-181b-5p mimic, the expression of p-Akt and VEGF were increased, while the expression of ES was decreased compared with levels in the control OGD-induced BMVECs. In contrast, treatment with an inhibitor of miR-181b stimulated PTEN expression and inhibited the Akt pathway, with increased ES and decreased VEGF levels (Figure 5A–C). Our results suggest that miR-181b-5p reduces PTEN, which activates the Akt pathway, leading to upregulated expression of VEGF and reduced expression of ES, thus stimulating angiogenesis in OGDinduced BMVECs.
We also determined the expression levels of apoptosis-related molecules, cleaved caspase-3/8/9, Bcl-2, and Bax. The levels of cleaved caspase-3/8/9 and Bax proteins were significantly downregulated in OGD-induced BMVECs after transfection with miR-181b-5p mimic, while Bcl-2 was upregulated (Figure 5A, D and E). In OGD-induced BMVECs transfected with the miR-181b-5p inhibitor, the levels of cleaved caspase-3/8/9, Bax, and Bcl-2 proteins showed opposite changes. These results further confirmed that miR-181b impeded OGD-induced apoptosis in BMVECs, which was consistent with the flow cytometry analysis in Figure 3B.
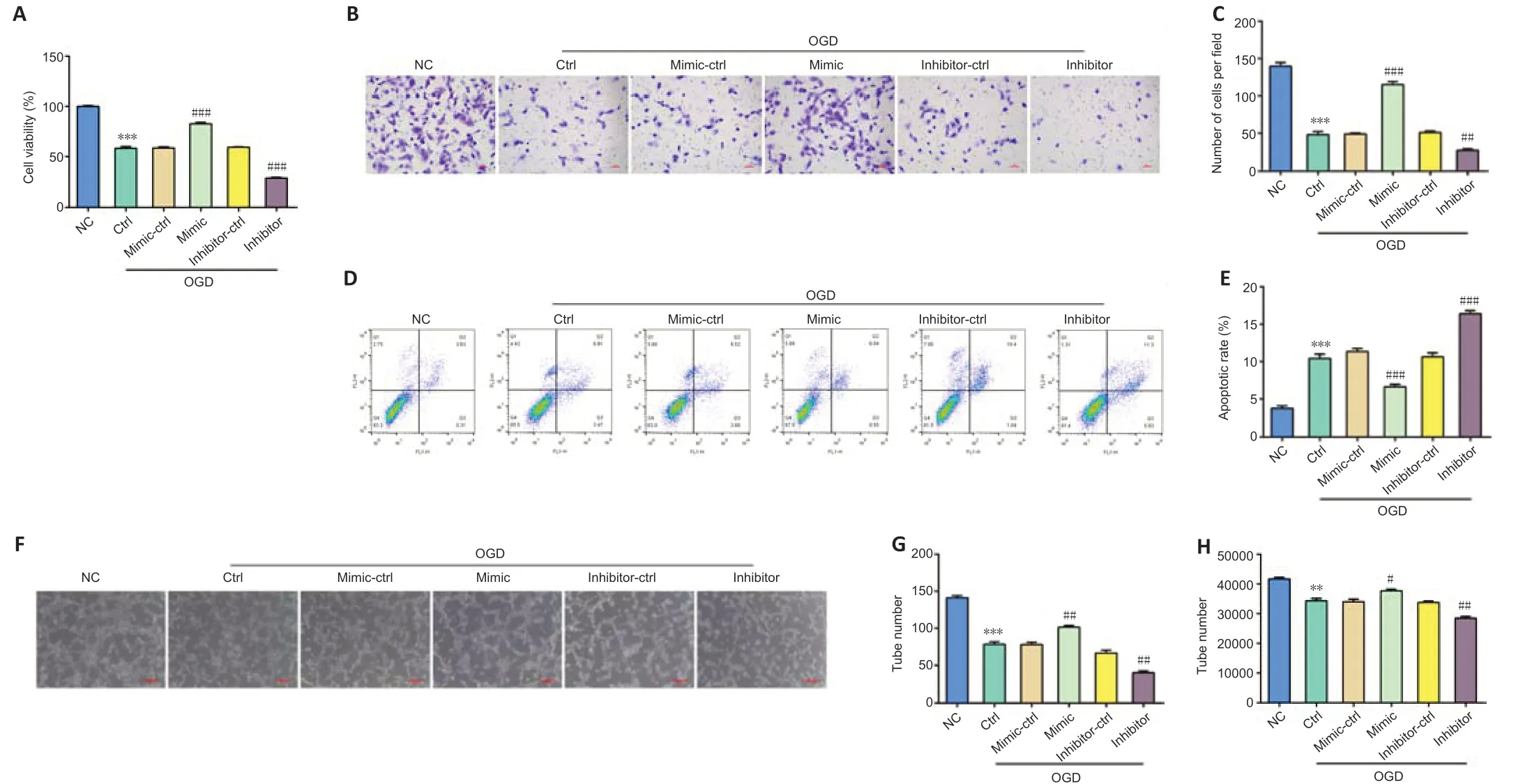
Figure 3|miR-181b promotes the cell proliferation, migration, and angiogenesis in OGD-induced BMVECs.
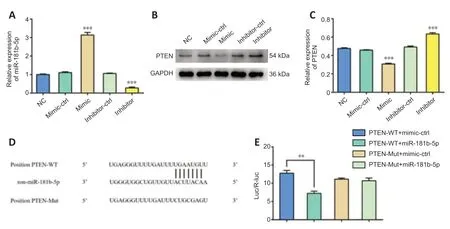
Figure 4|miR-181b suppresses PTEN expression via binding to the 3'-UTR of PTEN mRNA.
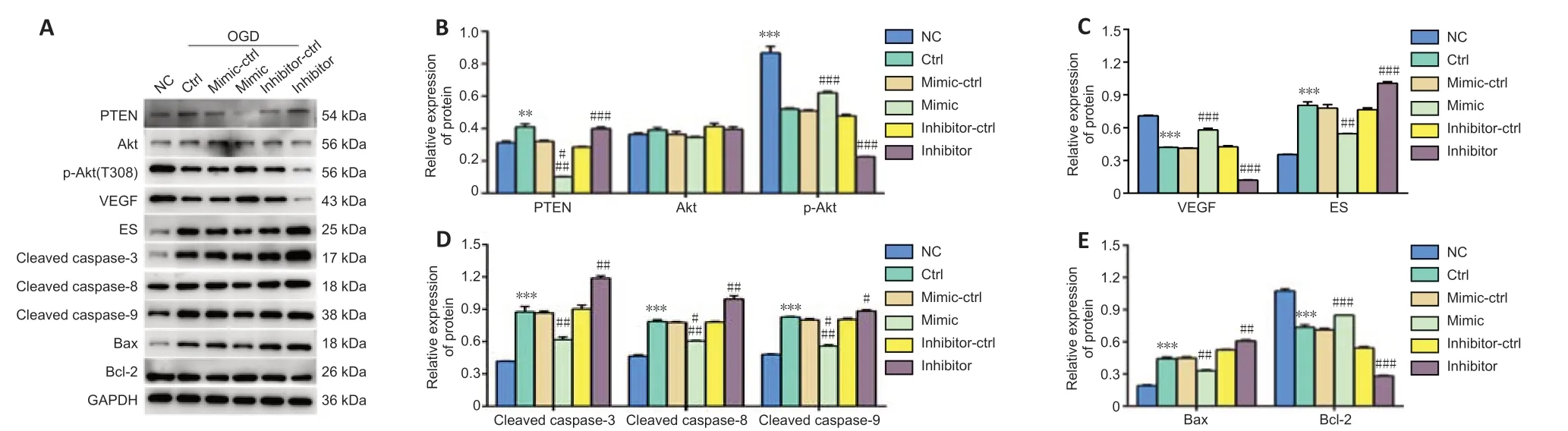
Figure 5|Changes in angiogenesis and apoptosis-related proteins in OGD-induced BMVECs under miR-181b treatment.
Discussion
Post-IS collateral circulation may modify the effect of neuroprotective therapies, highlighting the importance of the early establishment of collateral circulation in the treatment of IS. In our previous neuroprotection trial, butylphthalide, which improves regional microcirculation and increases the blood flow of ischemic brain tissue, was found to be more effective than neurotrophic and neuroprotective drugs (Xue et al., 2016). Angiogenesis plays an important role in the repair and regeneration of brain damaged by ischemia, and the recovery of blood flow increases the delivery of oxygen and nutrients to injured brain tissues (Arenillas et al., 2007; Yin et al., 2015). Angiogenesis is strictly regulated under various pathological conditions. Boosting angiogenesis by targeting various angiogenic factors may be a useful treatment for IS. Angiogenesis is modulated by several pro-angiogenic and anti-angiogenic factors, such as VEGF and ES, which we previously confirmed to be involved in endothelial progenitor cell proliferation in the early phase of IS (Xue et al., 2017). Over the last decade, miRNAs have been found to participate in the regulation of angiogenesis and play a bidirectional role in angiogenesis by targeting various genes. miR-132 diminishes cerebral injury by suppressing matrix metallopeptidase 9, which plays a major role in blood-brain-barrier disruption (Zuo et al., 2019). miR-126 represses sprouty-related EVH1 domain-containing protein 1 and phosphoinositide-3-kinase regulatory subunit 2, leading to activation of the VEGF pathway (Fish et al., 2008). miR-23 and miR-27 boost angiogenesis by targeting Sprouty2 and Sema6A, which exhibit anti-angiogenic activity (Zhou et al., 2011). miR-181b is one of the downregulated miRNAs that were identified in the brains of hypoxic-preconditioning-induced or MCAO-induced mice (Liu et al., 2012), and miR-181b has been reported to be involved in the regulation of angiogenesis under hypoxic conditions (Zhang et al., 2020). A recent study suggested that miR-181b suppresses angiogenesis by targeting cellular communication network factor 1 to inhibit the 5′-adenosine-monophosphateactivated protein kinase signaling pathway in hypoxia-stimulated endothelial cells and a hindlimb ischemia model (Li et al., 2021). However, the effects of miR-181b on IS remain controversial. A clinical risk study of single nucleotide polymorphisms indicated that miR-181b polymorphisms may singly or jointly contribute to the risk of IS (Han et al., 2018). Suppression of miR-181b, which targets heat shock protein family A member 5 and ubiquitin carboxylterminal hydrolase isozyme L1, induced neuroprotection in MCAO-induced micein vivo, and downregulation of miRNA-181b protects the mouse brain from IS and against hypoxic injury in N2A cells by repressing nuclear factor kappa-B signaling pathways (Peng et al., 2013). However, another study demonstrated that electroacupuncture induced high levels of miR-181b in the penumbras and improved neurobehavioral function rehabilitation in rats by regulating epigenetic changes to directly act on miR-181b and its downstream genes (Deng et al., 2016). We also identified a neuroprotective function and mechanism of miR-181b in IS in this study. Our results indicated that overexpression of miR-181b through administration of miR-181b-agomir into rats rescued nervous system function and reduced infarction volume in an MCAO model. Inhibition of miR-181b in rats through injection of miR-181b-antagomir damaged brain tissues much more than that observed in the MCAO model group, which showed a higher score and a larger infarction area. We also detected higher CD31-positive rates in miR-181b-agomirtreated MCAO rats compared with agomir-Ctrl group, which implied that miR-181b protects neurological function by inducing angiogenesis. To further explore how miR-181b regulates post-stroke angiogenesis, we constructed an OGD model using BMVECs. Overexpression of miR-181b promoted cell proliferation, migration, and angiogenesis and decreased apoptosis in OGDinduced BMVECs. Inhibition of miR-181b resulted in the opposite phenotypes. These results confirmed that miR-181b plays a neuroprotective role in IS by promoting angiogenesis, which is consistent with the results of a previous study. Exosomal miR-181b-5p also showed the same promotion of migration distance and tube length in BMVECs after OGD (Yang et al., 2018).
miRNAs play important roles in various biological processes by binding to different target genes. We searched for target genes of miR-181b in angiogenesis using Starbase and miRDB. PTEN is a crucial mediator of angiogenesis in the inactivation of the PI3K/Akt pathway and was predicted to be a miR-181b target gene. As a major negative regulator of Akt activation, PTEN has been confirmed to be involved in the activation of Akt signaling after cerebral ischemia and TBI (Guo et al., 2013; Xue et al., 2018; Chen et al., 2020). We verified that miR-181b bound to the 3′-UTR of PTEN mRNA and reduced PTEN levels, and binding to the 3′-UTR of PTEN was required for this effect. In OGD-induced BMVECs treated with miR-181b overexpression, the Akt pathway was activated, leading to increased VEGF and decreased ES, and promoted angiogenesis after IS. This finding is similar to that of a previous study reporting that miR181b-containing exosomes facilitate poststroke angiogenesis by upregulating the expression of HIF-1α and VEGF and downregulating the protein expression of tissue inhibitors (Yang et al., 2018). Furthermore, a decrease in cleaved caspase-3/8/9 and Bax, induced by miR-181b overexpression, was found to suppress apoptosis and promote proliferation of OGD-induced BMVECs. However, the exact molecular mechanism through which miR-181b influences these proteins remains unknown. Notably, angiogenesis therapy through the inhibition of PTEN may also have the capacity to induce a tumorigenic response. Thus, further studies to investigate the reduction in these effects and tests on the safety of miRNAs are urgently needed for stroke therapy.
This study has some limitations. In addition to angiogenesis, neurogenesis, synaptogenesis, and axonal remodeling also play important roles in promoting neurological recovery, so there may be other mechanisms that could account for how miR-181b exerts neuroprotective effects. This remains to be clarified in future studies. Additionally, we did not conduct a knock-out validation to test the effects of blocking PI3K/Akt signal pathway on angiogenesis using PI3K or Akt inhibitor. Finally, whether other signal pathways also contribute to proangiogenesis effects following IS needs further exploration.
In conclusion, we found that miR-181b promoted angiogenesis in IS by activating the Akt pathway, leading to the regulation of VEGF and ES by binding to PTEN. These results suggest that the miR-181b/PTEN axis might be a novel therapeutic target for IS. The application of exogenous agomir-181b may be a potential strategy for inducing angiogenesis in neurological and cerebrovascular diseases.
Author contributions:Study design: LXX, HC; experiment implementation: LXX, LYS; data analysis: LXX, LYS, HMW; literature review: KLL, LGH, JYX; manuscript writing: LX X, LYS, HC; manuscript review: ZG, YWZ, HC. All authors read and approved the final version of the manuscript.
Conflicts of interest:The authors declare that they have no competing interests.
Data availability statement:All relevant data are within the paper and its Additional files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Julien Rossignol, Central Michigan University, USA; María José Pérez-Alvarez, Autonomous University of Madrid, Spain; Noela Rodriguez-Losada, University of Malaga, Spain.
Additional files:
Additional Figure 1: The maps of PTEN and psiCHECK™-2 plasmid.
Additional Figure 2: BMVEC has a high CD31-positive rate.
Additional file 1:Open peer review reports 1–3.

- 中国神经再生研究(英文版)的其它文章
- Mesenchymal stem cells, extracellular vesicles, and transcranial magnetic stimulation for ferroptosis after spinal cord injury
- Inducing prion protein shedding as a neuroprotective and regenerative approach in pathological conditions of the brain: from theory to facts
- Use of mesenchymal stem cell therapy in COVID-19 related strokes
- Brain organoids are new tool for drug screening of neurological diseases
- Emerging roles of astrocytes in blood-brain barrier disruption upon amyloid-beta insults in Alzheimer’s disease
- External anal sphincter electromyography in multiple system atrophy: implications for diagnosis, clinical correlations, and novel insights into prognosis