
Emerging roles of astrocytes in blood-brain barrier disruption upon amyloid-beta insults in Alzheimer’s disease
2023-02-13 12:41QianYueMaggiePuiManHoi
中国神经再生研究(英文版) 2023年9期
Qian Yue , Maggie Pui Man Hoi ,
Abstract Blood-brain barrier disruption occurs in the early stages of Alzheimer’s disease. Recent studies indicate a link between blood-brain barrier dysfunction and cognitive decline and might accelerate Alzheimer’s disease progression. Astrocytes are the most abundant glial cells in the central nervous system with important roles in the structural and functional maintenance of the blood-brain barrier. For example, astrocytic coverage around endothelial cells with perivascular endfeet and secretion of homeostatic soluble factors are two major underlying mechanisms of astrocytic physiological functions. Astrocyte activation is often observed in Alzheimer’s disease patients, with astrocytes expressing a high level of glial fibrillary acid protein detected around amyloid-beta plaque with the elevated phagocytic ability for amyloid-beta. Structural alterations in Alzheimer’s disease astrocytes including swollen endfeet, somata shrinkage and possess loss contribute to disruption in vascular integrity at capillary and arterioles levels. In addition, Alzheimer’s disease astrocytes are skewed into proinflammatory and oxidative profiles with increased secretions of vasoactive mediators inducing endothelial junction disruption and immune cell infiltration. In this review, we summarize the findings of existing literature on the relevance of astrocyte alteration in response to amyloid pathology in the context of blood-brain barrier dysfunction. First, we briefly describe the physiological roles of astrocytes in blood-brain barrier maintenance. Then, we review the clinical evidence of astrocyte pathology in Alzheimer’s disease patients and the preclinical evidence in animal and cellular models. We further discuss the structural changes of blood-brain barrier that correlates with Alzheimer’s disease astrocyte. Finally, we evaluate the roles of soluble factors secreted by Alzheimer’s disease astrocytes, providing potential molecular mechanisms underlying blood-brain barrier modulation. We conclude with a perspective on investigating the therapeutic potential of targeting astrocytes for blood-brain barrier protection in Alzheimer’s disease. Key Words: Alzheimer’s disease; amyloid-beta; astrocyte (astroglial)-endothelial interaction; astrocyte pathology; blood-brain barrier; blood-brain barrier disruption; brain endothelial cell; neuroinflammation; reactive astrocyte
Introduction
Astrocytes (also known as astroglia) are the most abundant types of glial cells in the central nervous system (CNS). Under normal physiological conditions, astrocytes exhibit an intricate spongiform morphology (classically depicted as a star-like appearance), protruding their fine processes to make millions of contacts with neuronal synapses and vascular capillaries to modulate vital functions and maintain homeostasis (Schiweck et al., 2018). Besides regulating neuronal functions (via mechanisms such as synaptic pruning, neurotransmitter clearance, and extracellular K+buffering), astrocytes serve as an indispensable cell type in the neurovascular unit, which is the fundamental cellular unit of the blood-brain barrier (BBB). In neurovascular unit, astrocytes ensheath capillary endothelial cells and pericytes with endfeet and cover almost the entire cerebrovascular surface. The triad of brain endothelial cells, pericytes, and astrocytes in the highly specialized perivascular structure are continuously communicating with each other for promoting the stabilization, maintenance, and immune quiescence of the BBB (Zhao et al., 2015).
Increasing evidence supports that BBB alteration is a prodromal contributing factor for disease progression and possible pathogenesis in various sporadic neurodegenerative disorders such as late-onset Alzheimer’s disease (AD), as early cerebrovascular dysfunction precedes neuronal atrophy and cognitive decline (Sweeney et al., 2019). Impaired BBB in AD displays increased permeability, impaired function of carriers or transporter proteins and degenerated neurovascular unit cell members (Sweeney et al., 2018). Increasing findings in AD cases have also pointed out that dysregulation of the interaction between endothelial and perivascular cells (e.g., pericytes (Halliday et al., 2016), astrocytes (Abbott et al., 2006)) is involved in the progression of BBB breakdown. In this review, we focus on discussing the relevance of astrocyte alteration in response to amyloid pathology in the context of BBB dysfunction.
As part of CNS defensive mechanisms, astrocytes become reactive when they are challenged by various kinds of insults, including viral and bacterial infections and aggregation-prone proteins such as α-synuclein, transactive response DNA binding protein of 43 kDa (TDP43) and huntingtin. BBB breakdown in AD is possibly due to amyloid-beta (Aβ), tau, as well as neuroinflammation, oxidative stress, and apolipoprotein E allele 4 (APOE4) (Erickson and Banks, 2013). Reactive astrocytes are identified as a component of senile plaque in the cortex of AD patients, infiltrating and enwrapping Aβ deposits with their processes. Astrocytes respond to Aβ plaques and astrogliosis (reactive astrocytes) are thus induced (Perez-Nievas and Serrano-Pozo, 2018) and exhibit typical characteristics (such as hypertrophy, abnormal glutamate homeostasis, and aberrant energy metabolism) (Perez-Nievas and Serrano-Pozo, 2018). However, the role of Aβ-mediated reactive astrocytes in AD pathophysiology, particularly the astroglial-endothelial interaction under this pathological environment, is still not well understood. Here we review evidence from human pathology, animal, and cellular models of AD to further apprehend the intertwined relationship between astrocytes and brain endothelial cells in the presence of amyloid pathology.
Search Strategy and Selection Criteria
The articles cited in the present review were searched on Web of Science, Google Scholar, and PubMed databases with key search terms including astrocyte, brain endothelial cell, Alzheimer’s disease, amyloid-beta, and blood-brain barrier. The search mainly encompassed papers published from 2010 to 2022. Earlier original articles of the first scientific discovery were also cited.
Role of Astrocytes in the Physiology of the Blood-Brain Barrier
Structural support
Astrocytes have been divided into two main subtypes, protoplasmic or fibrous according to morphologies and anatomical locations. Protoplasmic astrocytes are predominantly involved in the coupling between astrocytes and brain capillaries (Verkhratsky et al., 2018). Mature astrocytes appear in a starlike shape, consisting of cell somata, processes with myriad branchlets, and endfeet. At the capillary level, BBB is comprised of an intricate multicellular structure, where astrocytes extend endfeet that ensheath capillary blood vessels and pericytes. At arteriole and vein levels, perivascular astrocytes make direct contact with the vasculature via their cell somata (Bardehle et al., 2013), where astrocytic endfeet also form an additional barrier called glial limitans that exclude blood-borne factors (Horng et al., 2017). The coverage of astrocytic endfeet or somata around vessels maintains the BBB integrity and the normal morphology of astrocytes is essential for the tightness of additional astrocytic coverage.
The stable contact between blood vessels and astrocyte endfeet depends on their adhesion to basal lamina via ligand-receptor interaction. α, β dystroglycan expressed both in brain microvessels and astrocyte endfeet facilitate their adherence to the vascular basal lamina (laminins). In addition, Integrin β1 on microvessels and integrin α6β4 on astrocyte endfeet were also reported to mediate their binding with basal lamina (laminins) (Milner et al., 2008). In addition, substance exchange between astrocytes and endothelial cells including water, ions, nutrients, or brain-derived wasteful molecules mainly occurs in the area between astrocytic endfeet and blood vessels (Cohen-Salmon et al., 2021).
The polarity of endfeet caused by the abundant distribution of water channel aquaporin 4 (AQP4) and gap junctions such as connexin 43 (Cx43) is important for endfeet structure and endothelial-astrocyte communication (Cohen-Salmon et al., 2021). There is a relationship between AQP4 and Cx43 that perivascular astrocytic AQP4 expression is proportional to Cx43 expression level, and coverage of endfeet is parallel with Cx43 expression level (Cibelli et al., 2021). AQP4 can be anchored by scaffold molecules such as syntrophin to the astrocyte endfeet membrane, which is important in maintaining endfeet structure and polarity (Nagelhus and Ottersen, 2013).
Biochemical support
Astrocytes promote BBB integrity partly by enveloping brain vessels with endfeet, and partly by releasing a series of promotive factors, including glial cell line-derived neurotrophic factor (GDNF), transforming growth factor-β (TGFβ), basic fibroblast growth factor (bFGF) and angiopoietin 1 (ANG1) (Figure 1) to maintain BBB integrity. Astrocytic GDNF binds to endothelial GDNF family receptor α1 and RET receptor kinase. Downstream signaling involves the activation of sirtuin 1/endothelial nitric oxide synthase (eNOS), phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), cyclic adenosine monophosphate (cAMP)/protein kinase A and inactivation of p38 mitogenactivated protein kinase (p38 MAPK) (Liu et al., 2022), which are involved in preventing tight junction (TJ)-related protein degradation. bFGF binds to endothelial FGF receptor 1 and activates sphingosine 1 phosphate receptor 1 (S1PR1). Suppression of RhoA, activation of ERK, and PI3K/AKT/Rac-1 in brain endothelial cells are involved in stabilizing TJ and adherence junction (AJ) proteins and attenuating inflammatory responses by bFGF (Wang et al., 2016). bFGF also upregulates Cx43, the loss of which has been suggested in AD cases (Xing et al., 2019). Astrocytic FGF-2 also stabilizes TJ- and AJ-related proteins (Alvarez et al., 2013). TGF-β exerts effects via activin receptor-like kinase 1 (ALK1) or ALK5. TGF-β upregulates gene expression of ZO-1 via ALK5/SMAD family member 2/3 (Smad2/3) mediated, Gli2-dependent manner (Fu et al., 2021) accompanied with increased activity of P-glycoprotein (P-gp) and decreased leukocyte infiltration. BBB dysfunction could in turn activate the TGF-β1 pathway in astrocytes (TGF-β1/Smad-2), which thereafter contribute to neuronal dysfunction (Senatorov Jr et al., 2019). ANG1 promotes vascular stabilization and neovascularization effects via the angiopoietin-1 receptor (also known as TIE2 or CD202B), which is an endothelium-specific receptor tyrosine kinase. ERK/ Ras-related C3 botulinum toxin substrate 1 (Rac1) is involved in ANG1/TIE2-mediated cell proliferation, migration, and angiogenesis (Fukuhara et al., 2009). Activation of PI3K /AKT-myocyte enhancer factor-2 (MEF2)-Krüppel-like factor 2 (KLF2) signaling is involved in vascular stabilization (TJ and AJ) and anti-inflammatory effects of ANG1.

Figure 1| Roles of astrocyte-derived growth factors in maintaining BBB integrity.
Astrocytes-derived sonic hedgehog (SHH) promotes expression of TJrelated proteins through activating endothelial receptor Patched 1 (PTCH1) and smoothened (SMO), therefore inducing subsequent GLI1 nuclear translocation (Alvarez et al., 2013; Lospinoso Severini et al., 2020). Besides, astrocytic SHH inhibits adhesion, migration, activation, and immune responses of T cells (Th1, Th17) by downregulating intercellular adhesion molecule 1 (ICAM-1) and chemokine (C-C motif) ligand 2 (CCL2) (Brambilla, 2019). In astrocytes stimulated by ischemic insults, SHH also leads to the upregulation of angiogenic growth factors such as ANG-1 mediated by nuclear receptor subfamily 2 group F member 2 (NR2F2) (Li et al., 2013). Production of astrocytic SHH could be inhibited by interleukin 1β (IL-1β) (Wang et al., 2014). The WNT/β-catenin pathway is generally acknowledged for stabilizing endothelial junctions and transporters. Astrocyte-derived WNT factor is crucial for BBB maintenance. Aging-associated loss of astrocytic WNT release progressively impairs endfoot integrity (Guérit et al., 2021). Astrocyte coculture upregulates the WNT family members WNT7A and WNT7B and the nuclear translocation of β-catenin, which stimulates the gene expressions of Claudin-5 and Occludin (Song et al., 2021). Astrocytic release of WNT factors as well as non-WNT-related ligand Norrin activates endothelial WNT/β-catenin activity (Guérit et al., 2021). It has been suggested that unbalance between SHH and WNT pathways is involved in BBB leakiness and neuroinflammation (Gozal et al., 2021). Astrocytes also secrete angiotensin-converting enzyme-1, by which angiotensin I is converted into angiotensin II. Angiotensin II interacts with endothelial type 1 angiotensin receptor (AT1) stabilizing junctional proteins (Alvarez et al., 2013). In addition, astrocyte-derived retinoic acid (RA) is identified to protect barrier properties by upregulating TJ proteins ZO-1 (Kong et al., 2015) and transporter proteins (P-gp, glucose transporter 1) (El Hafny et al., 1997), as well as attenuating endothelial inflammation (ICAM-1, vascular cell adhesion molecule 1) against inflammatory insults probably via activating antioxidant nuclear factor erythroid 2-related factor 2 (NRF2) pathway (Lippmann et al., 2014; Mizee et al., 2014). Increased RA is produced due to activated retinaldehyde dehydrogenase 2 (RALDH2) in reactive astrocytes and interacts with retinoic acid receptors in endothelial cells (Michinaga and Koyama, 2019). BBB inducement effect of RA is also in concert with other promotive pathways, such as SHH and WNT (Halilagic et al., 2007; Bonney et al., 2016).
Clinical Evidence of Astrocyte Alterations in Alzheimer’s Disease Patients
Astrocyte pathology in AD patients
Proinflammatory and oxidative profiles
Postmortem studies of AD patients have shown the co-localization of Aβ plaques with activated astrocytes indicated by increased glial fibrillary acid protein (GFAP) immunoreactivity (Bellaver et al., 2021). Astrocytic activation is usually viewed as an index of gliosis or a slow-developing process correlate of neural damage. In the clinic, a series of inflammatory factors such as S100 calcium binding protein (S100B), human cartilage glycoprotein 39 (HC-gp39) (also known as YKL40 and CHI3L1) (Bellaver et al., 2021), breast regression protein 39 (BRP-39) (Connolly et al., 2022), 18 kDa translocator protein (TSPO) (Tournier et al., 2020) and D serine (Balu et al., 2019) in cerebrospinal fluid (CSF) have been used to evaluate astrocyte reactivity, which are increased in AD patients. In addition, plasma levels of GFAP and S100B, have been used to indicate astrocyte reactivity (Brambilla, 2019). In astrocyte-derived exosomes of AD patients, there are high levels of complement effectors such as C1q, C3b, C3d, C4b, factor B, factor D, Bb and C5b-C9 terminal complement complex (Winston et al., 2019), as well as elevated levels of inflammatory cytokines such as tumor necrosis factor-α (TNF-α), IL-1β, IL-6, IL-8, IL-18, IL-23, IL-1 receptor 1 (Kaur et al., 2019) and chemokines CCL2, CCL3, CXCL-10, CXCL-12 and ICAM-1 (Viejo et al., 2022). In addition, astrocytes in AD patients display increased oxidative stress (Fracassi et al., 2021), increased level of advanced glycation end products (AGEs) (Chambers et al., 2020), and receptor for AGEs (RAGE) (Prasad, 2019). However, contradictory findings are also reported that antioxidant enzymes such as superoxide dismutase are suppressed (Fracassi et al., 2021), while heme oxygenase 1 is elevated in astrocytes in AD patients (Fernández-Mendívil et al., 2020).
Imaging evidence
Positron emission tomography (PET) has detected the alteration of astrocyte reactivity in AD patients. [11C]-deuterium-L-deprenyl ([11C]-DED) PET, and [11C]-BU99008 PET specifically target reactive astrocytes through binding to MAO-B and imidazoline 2 binding sites (I2-BSs) that are expressed on astrocytic mitochondrial membranes. Increased uptake of [11C]-DED and [11C]-BU99008, along with increased [11C]-PIB signals have been observed in the AD brain (Edison et al., 2018). Elevated uptake of acetate due to increased monocarboxylate transporter is predominantly observed in AD astrocytes associated with upregulated glycolysis. Increased uptake of acetate tracer, [11C]-acetate has been detected in mild cognitive impairment with an association with Aβ plaque (Duong et al., 2021). In addition, [11C]-PBR28 and CE-180 bind to TSPO and are usually used for detecting glial activation and neuroinflammation, however, it is not specific for astrocyte activation, and could also detect microglial inflammation. TSPO intensifies as AD proceeds and TSPO elevation in astrocytes precedes in microglia (Tournier et al., 2020).
Energy metabolism
Reduction of [18F]-FDG-PET, indicating glucose hypometabolism has been observed in Aβ positive districts in AD patients (Livingston et al., 2022). Glucose uptake by astrocytes is reduced in AD patients, characterized by downregulated levels of glucose transporters (GLUT1 and GLUT3) (Simpson et al., 1994). However, possibly acting as a compensatory mechanism to mitochondrial dysfunction and glucose transporters reduction, an overall upregulation of glycolytic enzymes (pyruvate kinase and lactate dehydrogenase) is observed as the disease progresses (Mulica et al., 2021). Moreover, glucose-6-phosphate dehydrogenase (G6PD) which has an important role in energy balance and is associated with neuroprotection, is observed to be reduced in AD astrocytes (van Gijsel-Bonnello et al., 2017).
Glutamate homeostasis
Glutamate homeostasis in the CNS is partly modulated by astrocytes via glutamate uptake or release. Impairments in the ability of astrocytes to uptake glutamate due to astrogliosis are hypothesized to be an early event in AD. Glutamate uptake is significantly reduced in the astrocytes of AD patients, accompanied by decreased expression levels of glutamate transporter 1 and glutamate and aspartate transporter (Hoshi et al., 2018). In addition, metabotropic glutamate receptor 5 (mGluR5) mediates glutamate-related excitotoxicity and is upregulated in astrocytes adjacent to Aβ plaques (Kumar et al., 2015). Glutamate is metabolized into glutamine in astrocytes by glutamine synthetase (GS) and glutamine is essential for glutamate synthesis in neurons (Andersen et al., 2020). Downregulated and dysfunctional GS colocalized with Aβ plaques is also observed in astrocytes from AD patients, which might indicate disturbed glutamate homeostasis (Ben Haim et al., 2015). CSF levels of glutamate and glutamine are used as markers for the alteration in the glutamate-glutamine cycle and to indicate the progression of AD (Dejakaisaya et al., 2021). Astrocytes also reuptake γ-aminobutyric acid (GABA). Reduced expression of GABA transporter (GAT3) (Fuhrer et al., 2017) and inhibited GABA metabolism has been reported in AD patient brain and human induced pluripotent stem cell (iPSC)-derived AD astrocytes (Andersen et al., 2022). In the postmortem brains of AD patients, increased production and release of GABA are observed in reactive astrocytes. GABA was released through astrocytic bestrophin 1 and has been suggested to be associated with impaired neuronal functions (Jo et al., 2014).
Aβ clearance
Reactive astrocytes are usually observed around Aβ plaque, which might relate to the role of astrocytes in Aβ reuptake and degradation. Aβ can be phagocyted by astrocytes through various membrane receptors and eventually degraded by a series of enzymes. Low-density lipoprotein receptor, low-density lipoprotein receptor-related protein (LRP1 and LRP4) (Zhang et al., 2020a), α7 nicotinic acetylcholine receptor (α7nAChR) (Xu et al., 2021) and scavenger receptor class B member 1 (SCARB1) (Bai et al., 2021) on astrocytic membrane have been reported responsible for Aβ internalization. Genetic deletion of theLrp4gene (specifically expressed in astrocytes) in AD mouse model 5xFAD showed that astrocytic uptake of Aβ was impaired (Zhang et al., 2020a). In postmortem AD brain tissues, reduced LRP4 is observed (Zhang et al., 2020a). While some Aβ reuptake proteins, such as opsonin complement C3 (C3), and SCARB1 are increased in reactive astrocytes extracted from AD patients (Viejo et al., 2022). LRP-mediated uptake of Aβ requires binding to assisting proteins such as apolipoprotein E (ApoE) and apolipoprotein J (ApoJ), α2-macroglobulin and α1-antichymotrypsin (Preman et al., 2021). Astrocytes are the main source of ApoE production in the brain. Decreased release of ApoE is observed in reactive astrocytes surrounding Aβ plaque (Bales et al., 1997). Aβ degradation by astrocytes relies on a series of Aβ degrading enzymes, including neprilysin (NEP), matrix metalloproteases (MMP-2 and MMP-9), insulin-degrading enzyme (IDE) and endothelin-converting enzyme (Yamamoto et al., 2018). The levels of these enzymes are increased in astrocytes in postmortem AD brain tissues (Viejo et al., 2022). Moreover, increased levels of amyloid precursor protein (APP), β-secretase (BACE-1), and γ-secretases presenilin (PSEN1 and PSEN2) have also been observed in AD reactive astrocytes from post-mortem studies (Carter et al., 2019). Taken together, reduced levels of reuptake receptors, assisting proteins, and Aβ degrading enzymes, as well as increased Aβ in AD patients might indicate dysfunctional activity of astrocytes in eliminating Aβ burdens.
More interestingly, an astrocyte model derived from iPSCs extracted from AD patients with PSEN1 ΔE9 mutation also showed severe astrocyte pathology (Oksanen et al., 2017). These AD astrocytes manifest AD hallmarks, including increased Aβ production and decreased Aβ uptake, altered cytokine release (such as IL-2, IL-6, IL-10, GM-CSF), Ca2+leakage from the endoplasmic reticulum (ER), and altered metabolism with mitochondrial dysfunction characterized as increased oxidative stress and reduced lactate secretion.
Preclinical Evidence of Astrocyte Alterations Related to Amyloid-Beta
In animal models of AD
Astrocyte activation is observed in AD-related transgenic mouse models which express transgenes of various forms of human mutations (APP,PSEN1,MAPT) associated with familial AD, such as the mouse models Tg2576, 5×FAD, APP/PS1, and 3×Tg-AD. All these transgenic mouse models develop AD pathology including APP overproduction and Aβ deposition at early ages (6–8 months), while 3×Tg-AD also exhibits tau pathology at older ages (12 months). Notably, pronounced astrocyte activation is observed in the PS1V97L transgenic mouse model (developed as a specific tool for studying Aβ oligomers-related pathogenesis bearing humanPSEN1*V97Lmutation) (Wang et al., 2019a). In line with this, intracranial ventricular injection (Urrutia et al., 2017) or intrahippocampal injection of Aβ-induced astrocyte activation provides further evidence of astrocyte reactivity in response to Aβ overproduction (Wang et al., 2018c; Ramírez et al., 2019). Various molecular and secretary alterations were observed in the astrocytes of AD mouse models. The most commonly observed alterations are the coidentification of reactive astrocytes with upregulated GFAP in the vicinity of Aβ plaques in the hippocampus (age 6–8 months), such as in 3×Tg-AD (Boscia et al., 2017) and 5×FAD (Sompol et al., 2017). Intense immunoactivity of potassium channel Kv3.4 colocalizing with Aβ and GFAP immunofluorescence was also observed in Tg2576 mice, indicating a disturbance of astrocytic K+homeostasis coincided with astrocyte reactivity and Aβ accumulation (Boscia et al., 2017). Intracellular Ca2+level (Dematteis et al., 2020) is increased according to the proteomics studies of the hippocampal astrocytes from 3×Tg-AD mice. Astrocytes in AD animal models generally display proinflammatory phenotypes, with increased productions and secretions of cytokines, chemokines, reactive oxygen species (ROS), and nitric oxide (NO) (Jorda et al., 2020a). In hippocampal astrocytes derived from 3×Tg-AD and APP/PS1 mice, secretions of neurotoxic factors TGFβ2 and TGFβ3 are increased (Tapella et al., 2018).
Hypometabolism in astrocytes is also commonly observed in AD mouse models. Immortalized astrocytes from 3×Tg-AD exhibited hypometabolism manifested as reduced glycolysis and oxidative phosphorylation (Dematteis et al., 2020). Reduced energy provision for neurons is observed in 5×FAD astrocytes as indicated by reduced monocarboxylate transporter activity (Andersen et al., 2021). Hypometabolism might be partially caused by decreased levels of GLUT1 (glucose uptake) and monocarboxylate transporter 1 (lactate release), which was observed in arcAβ transgenic mice bearing human transgenesAPPSweandAPPArctic(Merlini et al., 2011). Disturbed glutamate homeostasis has been reported to be associated with Aβ deposition in AD animal models (Dejakaisaya et al., 2021). GS expression in astrocytes (6–9 months), as well as GS-positive astrocytes (1–9 months), were reduced in 3×Tg-AD mice, in parallel with Aβ plaque development (Kulijewicz-Nawrot et al., 2013). In agreement, GLT loss was observed in APP/PS1 mice (6 months) (Fan et al., 2018) and 5×FAD mice (6–8 months) (Sompol et al., 2017). Reduction of glutamate and aspartate transporter is also reported in the hippocampus and cortex of AβPP23 transgenic mice preceding plaque formation (Schallier et al., 2011).
5×FAD astrocytes (12–14 months) showed impaired reuptake of Aβ, possibly due to decreased levels of SCARB (Iram et al., 2016) and LRP4 (Zhang et al., 2020a). Increased levels of RAGE and intracellular APP and/or Aβ in the astrocytes of aged 3×Tg-AD (22–24 months) were reported (Chen et al., 2013), indicating the defensive mechanisms to clear Aβ by reactive astrocytes in turn possibly burden astrocytic functions as the disease progresses. Data on the alterations in protein degrading enzymes (IDE, NEP) levels were conflicting. An early study showed a reduction of hippocampal IDE in 3×Tg-AD mice (Caccamo et al., 2005), but another study in 3×Tg-AD (3–15 months), as well as APP/PS1 mice (3–18 months), reported no obvious alteration in IDE level and Aβ degradation (Stargardt et al., 2013). In contrast, other studies reported increased IDE and NEP in APP(Swe)/PSEN1(A246E) (11–17 months), APP/PS1 (9 months), and Tg2576 mice (16 months) (Leal et al., 2006; Vepsäläinen et al., 2008). More interestingly, in astrocytes derived from PS1V97L transgenic mice, increased Aβ oligomers production and spread of toxic Aβ assemblies were observed, suggesting AD astrocytes might be involved in toxic Aβ species production and spreading to other parts of the brain (Wang et al., 2019a).
In summary, results from the studies in AD-related transgenic mouse models with Aβ pathology indicate a close association between Aβ pathogenesis and astrocyte activation. In line with observations in AD patients, astrocytes in AD mice also display inflammatory profiles and functional abnormality in energy metabolism, glutamate homeostasis, and Aβ clearance.
In cellular models of AD
Inin vitrostudies, external Aβ is applied to cellular models of astrocytes to investigate the underlying mechanisms of Aβ-induced astrocyte activation in isolated physiologic and pathological states. Human Aβ1–42soluble peptides (monomer, oligomer, and aggregates) are the mostly used Aβ species in cellular models including primary astrocytes cultured from rat (Sprague-Dawley and Wistar) and mouse (BALB/c, C57BL/6J, and SJL/J) models, and human glial cell (HA 1800) and human glioblastoma cell (U 87 MG).In vitroastrocytes activated by Aβ1–42exhibit upregulation of GFAP and intracellular Ca2+ increase (Ramírez et al., 2019). A previous study also shows that astrocytes were skewed into proinflammatory and oxidative profiles with upregulated secretions of cytokines, chemokines, adhesion molecules, MMPs, ROS, and NO, while a concomitant downregulated secretion of antioxidative factors such as glutathione (Wang et al., 2019b). DNA and lipid damages induced by oxidative stress are also observed. Table 1 summarizes the astrocytic secretions upon Aβ stimulation in various modelsin vitro.

Table 1 |Astrocytic secretions in Aβ-induced cellular models in vitro
Reductions in energy metabolism were observed in cultured astrocytes from the AD mouse model (5xFAD), possibly caused by downregulated expressions of GLUT1, glyceraldehyde-3-phosphate dehydrogenase (GAPD), α-ketoglutarate dehydrogenase (α-KGDH) and succinate dehydrogenase (SDH) (van Gijsel-Bonnello et al., 2017). In primary rat astrocytes, human oligomer (hAβo) downregulated preproinsulin mRNA and insulin protein, which might suppress neuronal insulin signaling and reduce neuronal activity (Takano et al., 2018). hAβo also directly impaired glutamate homeostasis by causing the loss of GLT1 and impaired glutamate reuptake (Dejakaisaya et al., 2021). Recombinant human Aβ1–42was observed to bind to primary rat astrocytes and enhanced the deleterious clustering of mGlu5R, which might increase glutamate-mediated excitotoxicity in astrocytes (Shrivastava et al., 2013).
Exogenous treatment of hAβo was reported to directly induce astrocytic responsesin vitro. In primary human astrocyte culture, hAβo stimulated the upregulation of astrocytic Aβ endocytosis (Montoliu-Gaya et al., 2018). Heat shock protein B1 (HspB1) (also known as Hsp25/27), which is capable of binding and sequestering extracellular Aβ was also observed stimulated by hAβo in primary rodent astrocyte cultures (Nafar et al., 2016). hAβo stimulated an increase of P-gp but alterations of LRP1, IDE, and NEP were not significantin vitro(Batarseh et al., 2017). In agreement, It was observed that in human primary astrocytes, hAβo or fibrillar Aβ alone cannot affect Aβ reuptake and degradation, while cotreatment with amyloid-related proteins, such as C1q and ApoE induces an increased level of SCARB, IDE, and NEP (Mulder et al., 2012). Therefore, it seems that activated Aβ clearance activity in astrocytes around Aβ might require the stimulation of amyloid-related proteins, not just Aβ. Decreased secretion of ApoE exosomes is observed in Aβ1–42stimulated astrocytes (Abdullah et al., 2016). In agreement, aggregated human Aβ1–42is also found to inhibit secretion and lipidation of ApoE causing intracellular accumulation in primary human and mouse glial cells (Chernick et al., 2018). Knockdown of astrocytic LRP1 aggravated hAβo-induced toxicity in mouse primary astrocytes (He et al., 2020), suggesting aberrant astrocytic ApoE/LRP1 signaling might affect Aβ endocytosis.
In summary, results from Aβ-stimulatedin vitrocellular models also show astrocyte activation exhibiting inflammatory and oxidative profiles and Ca2+overload. Functional abnormality including glucose hypometabolism, decreased glutamate reuptake and mGlu5R-mediated glutamate excitotoxicity occurred in Aβ-stimulated astrocytes. Aβ clearance and degradation are also affected. Most evidence indicates extracellular Aβ stimulation upregulates astrocytic endocytosis of Aβ, however, contradictory alterations, such as decreased secretion of ApoE also occur, which might need further elucidation.
Molecular mechanisms involved in astrocyte alteration in AD
Figure 2 depicts the cellular and molecular domain patterns in astrocytes that are specifically affected in response to Aβ pathology in AD. RAGE, LRPs, Tolllike receptors (TLRs), and SCARB expressed on astrocytes have been identified for their abilities of binding to Aβ and mediating downstream signaling pathways in astrocytes (Fakhoury, 2018).

Figure 2|Specific patterns of astrocyte activation in Alzheimer’s disease (AD).
In vitroandin vivoevidence has indicated that astrocytic proinflammatory and oxidative profiles induced by Aβ partly result from the activation of nuclear factor κB (NF-κB) signaling that can be triggered by TLRs (Wang et al., 2018c) and RAGE (Ding et al., 2020a). The Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) pathway, a defensive protective mechanism in response to CNS injury, was also reported to promote astrocyte reactivity in animal models of AD (Haim et al., 2015). Activation of STAT3 is involved in hypertrophy (Choi et al., 2020), and increased vascular endothelial growth factor (VEGF) production via hypoxia-inducible factor 1 alpha (HIF1α) (Toral-Rios et al., 2020) in AD astrocytes. Signaling mediated by TGFβ, interferon γ, estrogen receptors α and IL-6/gp130 could also activate STAT3 to further mediate anti-inflammatory, antioxidative, and neurotrophic effects (Kwon and Koh, 2020). Suppressor of cytokine signaling 3 was involved in NF-κB- and STAT3-induced inflammatory responses (Haim et al., 2015). Activation of p38, glycogen synthase kinase-3 beta, and tyrosine-protein kinase Fyn was also reported to be involved in astrocyte activation in AD models (Kheiri et al., 2019). Activation of signaling pathways including NLR family pyrin domain containing 3 (NLRP3) /caspase-1/IL-1β (Ebrahimi et al., 2018; Kuwar et al., 2021), calcineurin (CN)/nuclear factor of activated T cell (NFAT) (Furman et al., 2012), and c-Jun N-terminal kinase (JNK)/c-Jun/activator protein 1 (Wang et al., 2018a) was also involved in inflammation in AD astrocytes. Aβ caused deacetylation of histone H3 by promoting the accumulation of histone deacetylases (HDAC1, HDAC2, HDAC5), triggering proinflammatory responses and reducing brain-derived neurotrophic factor production (Wang et al., 2019b).
As mentioned above, reactive astrocytes located around Aβ plaques display elevated phagocytic activity, manifested as upregulated phagocytic receptors (P-gp, SCARB), protein degrading enzymes (IDE, NEP), and intracellular inclusions of Aβ, which closely correlated with activated autophagy in astrocytes (Sung and Jimenez-Sanchez, 2020). Accordingly, In AD patients, the protein degradation system in astrocytes is boosted, manifested as increased markers, such as cathepsins B, beclin-1, HSPB1, E3 ubiquitin-protein ligases, and calpain (Viejo et al., 2022) It seems that astrocyte in the vicinity of Aβ plaque exhibit activated autophagy and phagocytic activity. In AD mice bearing mutated human APP genes, astrocytes with positive immunoreactivity of microtubule-associated protein 1A/1B-light chain 3 are also observed around Aβ plaque (Pomilio et al., 2016). However, conflicting finding is also observed that Aβ pathology also induced dysfunction of autophagy in astrocytes, manifested as light chain 3II reduction and p62 aggregation (Hong et al., 2019), which indicates there seems to be a blockage in the autophagylysosome pathway.
Aβ-induced astrocytes also exhibited hypometabolism, which was partly caused by mitochondrial apoptosis. Decreased adenosine 5′-triphosphate generation, abnormal mitochondrial membrane potential (MMP), and cardiopulmonary resuscitation activity along with increased Bcl-2 and caspase 3 activities were observed in apoptotic astrocytes (Yao et al., 2018). In human astrocytes, Aβ-induced loss of excitatory amino acid transporter 1 (EAAT1) (Glt1 in rodents) and EAAT2 (Glast in rodents) might relate to decreased levels of Akt (p-Ser473) and mTOR (p-Ser2448). In agreement, decreased expression of Glt1 induced by CN/NFAT led to GLT1 internalization and ubiquitination in rodents (Dejakaisaya et al., 2021).
Aberrant Ca2+signaling is also induced by extracellular Aβ and glutamate overload. Calcium uprise elevates various Ca2+-binding proteins including S100B, calcyclin (S100A6), calbindin (CALB1), calsenilin (KCNIP3), and calretinin (CALB2). Ca2+released from ER internal store was also observed in Aβ-induced astrocytes via ER stress and phospholipase C (PLC)/inositol 1, 4, 5-triphosphate (IP3)-mediated pathway (Alberdi et al., 2013). It is suggested the aberrant Ca2+could also be associated with the elevated expression and activities of Kv3.4, Kca3.1, and Kir6.2 channels which disturb ion homeostasis (Boscia et al., 2017). It was observed in astrocytes near Aβ plaques in APP/PS1 mice, Cx43 and Cx30 expressions were elevated and Cx43 hemichannels were chronically opened leading to astrocytic Ca2+influx and adenosine 5′-triphosphate and glutamate release, in turn further triggering astrocyte activation (Smit et al., 2021). Furthermore, in APP/PS1 mice with astrocytespecific Cx43 deficiency, there was a decrease in astrocyte reactivity and a reduction in neuronal damage, but it was not the result of a decrease in Aβ plaque (Aβ load was the same as in APP/PS1 Cx43-positive animals), indicating Cx43 was a key downstream effector of astrocytic response to Aβ.
Apart from direct effects on astrocytes, Aβ species are likely to induce astrocyte reactivity through stimulating other Aβ-reactive cells in the brain such as microglia (Sofroniew, 2015; Xie et al., 2020) and oligodendrocytes (Quintela-López et al., 2019; Ferreira et al., 2020).
Shortcomings of the current murine AD models
Similar findings about the alterations of astrocytes were reported in AD patients, or AD animal models and Aβ-induced cellular models, including proinflammatory and oxidative profiles, decreased glucose metabolism and glutamate reuptake, as well as increased Aβ clearance. However, there are some shortcomings in using murine AD models in AD research. Firstly, transgenic murine AD models (mTg-AD models) are widely applied in the preclinical studies of AD. Current mTg-AD models mostly mimic familial AD pathogenesis through being transfected in numerous AD risk mutations in human geneAPP,PSEN1,PSEN2, andMAPT. However, only 5% of AD is familial AD, which may indicate the findings observed in familial AD models have little contribution to AD therapy. The other 95% is sporadic AD, the pathogenesis of which is multifactorial (Riemens et al., 2020). Currently applied sporadic AD animal models reviewed in (Zhang et al., 2020b) mimic a series of pathologies, such as disturbed glucose metabolism, lipid metabolism, oxidative stress, and imbalanced metal ions by using specific stimuli such as streptozotocin, high cholesterol, D-galactose, and ions. Pathogenesis and disease progression in AD patients are very complicated. For example, reduced insulin and corresponding signaling pathway occur when human get old, which could increase AD risk (Gunn-Moore et al., 2018). Murine AD model was usually established by targeting certain pathologies, thus cannot fully mimic pathology in AD patients. It was reported that mouse strains can drift unpredictably after backcrossing, which might lead to massive loss of neurons (Ittner et al., 2008). In addition, AD murine models basically have the homogenous genetic backgrounds and short lifespans, which are different from humans and may affect the disease progression (Götz et al., 2018).
Structural Changes at the Blood-Brain Barrier that Correlate with Alzheimer’s Disease Astrocytes
In AD patients, BBB structural alterations and barrier leakages are observed in post-mortem brains and gadolinium-based dynamic contrast-enhanced MRI studies, manifested as disrupted expressions of junctional and transporter proteins accompanied by elevated oxidative stress and vascular inflammation. In line with this, reactive perivascular astrocytes with swollen endfeet and reduced astrocytic coverage around endothelial cells in deformed capillaries are often identified (Nehra et al., 2022). Oeckl et al. (2019) recently observed serum GFAP levels (a biomarker for astrocytic activation) were higher in AD patients when compared with normal control participants. In this regard, elevated serum GFAP levels in APP/PS1 mice have been reported to correlate with increased gadolinium perfusion in the brain (Scholtzova et al., 2008). Furthermore, hippocampal astrocytes or astrocytic extracellular vesicles isolated from 3×Tg AD mice failed to maintain normal TJ proteins expression in brain endothelial cells compared to those from wild-type mice (Kriaučiūnaitė et al., 2021). These findings might reflect the association between BBB breakdown and astrocyte activation.
Atrophic and hypertrophic astrocytes
BBB integrity is partly dependent on the structural coverage of astrocytes. The morphological alterations of astrocytes observed in CNS diseases include decreased cell volume, cell surface, and process numbers (atrophic features) along with endfeet retraction and detachment from blood vessels accompanying BBB breakdown (Steiner et al., 2012; Arranz and De Strooper, 2019). Such atrophic astrocytes are also detected in familial AD human postmortem brain tissues (Rodríguez-Arellano et al., 2016) and iPSCs-derived astrocytes from early-onset and late-onset familial AD patients (Jones et al., 2017). On the other hand, around Aβ plaques hypertrophic astrocytes with increased surface and volume were also detected, except from those regions in sub-pial and perivascular areas (Dickson et al., 1988).
In 3×Tg AD mice at pre-plaque stages, atrophic astrocytes have been observed in numerous brain districts such as the entorhinal cortex (1 month of age), prefrontal cortex (3 months of age), and hippocampi (CA1) (9–12 months) (Verkhratsky et al., 2016). In contrast, hypertrophic astrocytes were detected around Aβ plaques in 3×Tg AD mice at this later stage (Olabarria et al., 2010), These data indicated atrophic astrocytes occurring in perivascular areas with morphological shrinkage might contribute to their loss of supportive functions for BBB integrity.
Aquaporin 4, Connexin 43, and basal lamina
Specifically, marked redistribution of water channel AQP4 was observed in AD patients (Zeppenfeld et al., 2017). A similar phenomenon was also reported in AD mouse models demonstrating AQP4 proteins were reallocated from perivascular endfeet to the astrocytic processes surrounding Aβ plaques (Wilcock et al., 2009; Yang et al., 2011; Smith et al., 2019). Increased redistribution of AQP4 in the processes surrounding Aβ plaques is possibly related to upregulated Aβ clearance via AQP4, as deletion of AQP4 in APP/PS1 mice aggravated Aβ accumulation (Xu et al., 2015). The relocation of AQP4 indicated a loss of astrocytic polarity, leading to astrocyte edema and subsequent detachment of endfeet from endothelial basal lamina, and it is suggested signaling of Ca2+/calmodulin (CaM)/protein kinase A might be involved in the redistribution of AQP4 in astrocytes in edema-related pathology (Patabendige et al., 2021).
Besides, previous studies observed increased expression of astrocytic Cx43 around Aβ plaque in the postmortem brain tissues of AD patients (Nagy et al., 1996; Xing et al., 2019). In APP/PS1 mice, however, upregulated Cx43 expression was observed in a majority of plaques but Cx43 reduction was detected around newly formed plaques (Mei et al., 2010). Cibelli et al. (2021) have demonstrated Cx43 was abundantly distributed around brain vasculature, and knockout of Cx43 or its carboxy terminal domain led to BBB leakiness. However, altered Cx43 expression in astrocytic endfeet around blood vessels in AD-related models had not been reported so far.
In addition, an altered basement membrane in a capillary with a detachment of endfeet from basal lamina was indicated in AD studies. In a study using transgenic arcAβ mice, pronounced loss of dystroglycan expression in astrocytes and blood vessels occurred in parallel with vascular leakages and Aβ deposition (Merlini et al., 2011). AD astrocytes also release metalloproteinases (MMP-2, MMP-3, MMP-9) which are capable of proteolytically degrading dystroglycan, further contributing to endfeet detachment (Rempe et al., 2016).
Taken together, results from these studies suggested that the majority of perivascular astrocytes in AD have atrophic morphology with shrink cell somata and reduced possess number, possibly causing loosening of the barrier. In addition, the redistribution of key proteins expressed on astrocytic endfeet such as AQP4 and Cx43 lead to edema and swollen endfeet, along with increased MMPs release and decreased dystroglycan level, all contribute to astrocyte detachment in the capillary basement membrane affecting barrier integrity in AD.
Astrocytic Secretions in Alzheimer’s Disease Models and Their Potential Blood-Brain Barrier-Modulating Mechanisms
Detrimental factors
Besides structurally supporting the BBB with endfeet, astrocytes are highly secretory cells, which continuously regulate BBB stability by paracrine signaling. In AD, astrocytes shift to proinflammatory states secreting various signaling molecules including cytokines, chemokines, and growth factors, which play both neuroprotective and neurotoxic roles in brain lesions. However, AD-induced astrocytic secretions might contribute to BBB disruption possibly by loosening endothelial cell junctions and increased vascular inflammation, which is a physiological response during the progress of defensive inflammation, but in AD this defensive inflammation might turn into prolonged and pathogenic vascular leakages. In addition, decreased glutamate reuptake in AD astrocytes causing excessive extracellular glutamate, as well as decreased astrocytic secretion of ApoE, might also produce negative effects on the health of brain endothelial cells. Figures 3 and 4 depict the possible interconnected roles of AD astrocyte-derived secretions in inducing BBB breakdown. Next, we will focus on the specific signaling pathways that are related to vasoactive mediators secreted from reactive astrocytes, and their potential roles causing detrimental effects on BBB integrity and functions.
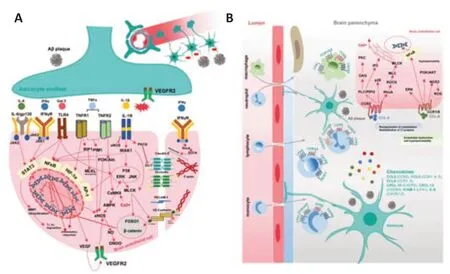
Figure 3|Potential roles of astrocyte-derived cytokines and chemokines stimulated by AD pathology in dissociating endothelial integrity and inducing vascular inflammation.

Figure 4|Potential roles of astrocyte-derived detrimental factors stimulated by AD pathology in dissociating endothelial integrity and inducing vascular inflammation.
Cytokines
In vitrostudies using astrocyte cellular models have shown that Aβ species stimulate astrocytes to release proinflammatory cytokines to various degrees, with soluble human Aβ oligomers being the most potent (Table 1). These cytokines include master regulators such as TNF-α (Urrutia et al., 2017), interleukins (IL-1β, IL-6) (Ebrahimi et al., 2018; Braidy et al., 2019; Ramírez et al., 2019) and IFNγ (Ramírez et al., 2019), which are expressed normally at very low levels but are induced rapidly in response to infections or insults. Aβ oligomers also upregulate cyclooxygenase-2 (Wang et al., 2018a), Galectin-3 (Ramírez et al., 2019), and IL-3 (McAlpine et al., 2021). They stimulate the activation of NF-κB signaling which further enhances the transcription of inflammatory cytokines. Simultaneously, the BBB TJs and AJ are under constant threat from these proinflammatory mediators. The BBB destabilizing effects of proinflammatory cytokines through signaling to NF-κB via protein kinase C (PKC) (Aveleira et al., 2010), PI3K/AKT (Min et al., 2007), receptorinteracting protein (Downes and Crack, 2010; Savard et al., 2015) and the Src family kinases Fyn/Lyn (Gong et al., 2008) under inflammatory conditions are well documented (Alvarez et al., 2013). The cytokines-mediated NFκB activation directly downregulates the gene expressions of TJs, AJs, and endothelial efflux transporters while activating endothelial inflammatory responses and inducing oxidative stress (Song et al., 2020). There is overplaying between WNT/β-catenin and NF-κB signaling. Endothelial β-catenin deficiency or downregulation of WNT/β-catenin signaling leads to decreased levels of TJs proteins and disruption of intercellular junctions, enhancing BBB destabilization and breakdown (Song et al., 2021; Hussain et al., 2022). Lamberti et al. (2001) have reported the essential role of inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ) in decreasing β-catenin transcriptional activation, thus downregulating β catenin-related genes in TNF-α-induced mouse embryo fibroblasts, while inhibitor of nuclear factor kappa-B kinase subunit alpha (IKKα) exerts opposite effects. The potential role of IKKβ and IKKα in regulating a panel of genes related to vascular cell inflammation, adhesion, and migration has been reported in cancer and vascular diseases such as diabetes and atherosclerosis (Gamble et al., 2012; Sui et al., 2014). Similar findings have not been reported in AD-induced BBB disruption. TNF receptor-1 (TNFR1) and TNFR2 are expressed in brain endothelial cells, and their activation by TNF-α and signaling downstream can activate the proviral integration site for Moloney murine leukemia virus-1 kinase, eNOS, and NO overproduction, which might cause endothelial disruption (Lu et al., 2020). Activation of Ca2+/Ca2+/calmodulin dependent protein kinase II (CaMKII)/ERK is reported in TNF-α-induced endothelial cells, which induce activation of MMP-9 and NF-κB signaling (Ding et al., 2019). In addition, activation of HIF1α/VEGF/VEGF receptor 2 (VEGFR2)/ERK pathway (Zhang et al., 2019) is found involved in the TNF-α-induced downregulation of TJ proteins. TNF-α also induces endothelial necroptosis through receptorinteracting protein 1 and mixed lineage kinase domain-like pseudokinase signaling (Chen et al., 2019). Astrocytic-derived IL-6 activates endothelial IL6R/gp130-mediated JAK2/STAT3 pathway, which induces multiple effects in brain endothelial cells including increased adhesion molecules, mitochondrial dysfunction, overproduction of ROS and NO, causing disruptions in endothelial functions (Valle et al., 2019). IL-6-induced TJs proteins (claudin-5 and ZO-1) reductions are mediated through MMP-induced degradation and ubiquitin proteasome-induced degradation. The effect of IL-1β is mediated via the activation of endothelial IL-1 receptor 1/IL-1 receptor associated kinase signaling (Ding et al., 2020b). IL-1β further induces oxidative stress, mitochondrial dysfunction, and NF-κB-mediated inflammatory responses, and at the same time decreased eNOS and endothelial NO production (Song et al., 2019). IL-1β triggers nuclear translocation of Forkhead Box O1 and β-catenin through activating non-muscle isoform of myosin lightchain kinase, thereafter, downregulates the gene expression of claudin-5, while reduces membrane ZO-1 by phosphorylating ZO-1 in PKCθ-dependent manner (Beard Jr et al., 2014). IL-1β in turn activates astrocytes and induces HIF1α-mediated overproduction of VEGF (Argaw et al., 2006). LPS-induced astrocyte-conditioned medium upregulates the expression level of IL6R, gp130, TNFR, and IL-1 receptor in BECs (Yue et al., 2022b). IFNγ disrupts BBB integrity by disassembling TJs proteins, and depolymerizing F-actin (Rahman et al., 2018) through activating type II IFN signal, including activation of Ras homolog family member A and Rho kinase (ROCK) (Bonney et al., 2019). Galectin-3 induces NF-κB-mediated inflammatory responses via TLR4-mediated activation of ERK/JNK and JAK2/STAT3 along with activation of MMP-9 (Tan et al., 2021; Figure 3A).
Chemokines
Increased production of chemokines is observed in reactive astrocytes. Chemokines modulate ongoing immune responses as they are involved in activating microglia and inducing BBB opening to recruit infiltrating immune cells to clear CNS insults such as Aβ. However, the recruited cells are not able to clear massive amounts of aggregated Aβ causing prolonged chemokines production. Thus, chemokines are also thought to promote chronic neuroinflammation in the late stage of preclinical AD and determine its transition to symptomatic AD (Strobel et al., 2015). Macrophages, neutrophils, lymphocytes, and monocytes have been reported involved in neuroinflammation. The combination between chemokines and chemokine receptors (CXCR and CCR) expressed on the immune cell surface mediates the infiltration of the immune cells. Given the above information, AD astrocytes produce increased CCL1, CCL8, CXCL-10, CXCL-12, ICAM-1, and IL-8. Figure 3B shows the corresponding receptors for each chemokine and the distribution of these receptors on immune cells. CCL2 and CCL8 could also induce endothelial hyperpermeability. CCL2-CCR2 axis induces endothelial cytoskeleton reorganization and membrane TJs protein internalization via signaling pathways including RhoA/ROCK/MLC/MLCK, PLC/phosphatidylinositol 4, 5-bisphosphate (PIP2) /diacylglycerol/PKC, PLC/PIP2/inositol 1,4,5-triphosphate and p38 MAPK (Keep et al., 2018). CCL8 interacts with CCR1 and/or CCR8 (Li et al., 2020) and causes endothelial dysfunction by activating ERK, PI3K/AKT, and NF-κB pathways and inducing ROS-mediated hyperpermeability through activating NADPH oxidase 2 (Xue et al., 2021). CXCL-10 interacts with CXCR3 and induces infiltration of antibody-secreting cells, T cells, and leukocytes into CNS and increases vascular activation (Sorensen et al., 2018). It is reported CXCL-10 cannot impair barrier integrity directly, but by triggering TNF production in astrocytes via the MAPK pathway (ERK, JNK, p38) (Wang et al., 2018b). In addition, in our previous study, we observed the astrocyte-cultured medium after LPS-stimulation was rich in CXCL-10, CCL7, and CCL2, and increased monocyte adhesion on brain endothelial cells (Yue et al., 2022b; Figure 3B).
S100B
Astrocytes have the highest expression of S100 calcium-binding protein B (S100B) in the brain. Increased S100B release is observed in 5xFAD mice following cytoplasmic Ca2+uprise in reactive astrocytes via CN/NFAT pathway (Sompol et al., 2017). It is suggested hyperactive astrocytic CN/NFAT signaling might couple vascular pathology to neurodegeneration. S100B induces BBB disruption by binding to RAGE expressed on endothelial cells, which enhances the activities of A disintegrin and metalloproteinase 17 (ADAM17) leading to impaired endothelial glycocalyx shedding and hyperpermeability (Zou et al., 2022). On the other hand, S100B activates astrocytic TLR2 and RAGE and upregulates astrocytic VEGFA release (Ding et al., 2021; Figure 4A).
Vasoactive mediators
Vasoactive mediators including endothelin-1 (ET-1), arachidonic acids, and arachidonic acids metabolites (prostaglandin E2 (PGE2), epoxyeicosatrienoic acids, and 20 hydroxyeicosatetraenoic acid (20-HETE)) are upregulated in AD astrocytes via phospholipases A2 activation (Viejo et al., 2022). PGE2 and epoxyeicosatrienoic acids dilate blood vessels, whereas 20-HETE and ET-1 constrict. The activities of PGE2 are mediated through prostanoid receptors (EP1, EP2, EP3, and EP4). It was shown low PGE2 concentration induces vasodilation via EP4, but high PGE2 concentration constricts brain vessels via EP1, suggesting increased PGE2 production from reactive astrocytes leads to a reduction of cerebral blood flow (Czigler et al., 2020; Figure 4D). Under normal physiology astrocytes contribute to generating vascular tone by the tonic release of 20-HETE to act on vascular smooth muscle cells, thus it is speculated in AD reactive astrocyte compromise 20-HETE regulation disturbing capillary blood flow (Hall et al., 2014). In addition, studies have verified PGE2 could stimulate Ca2+uprise in capillary endothelial cells, partly by opening transient receptor potential vanilloid 4 (TRPV4) channel and partly through triggering ER Ca2+release (Harraz et al., 2018) through depleting PIP2, which is also involved in cerebral blood flow modulation. Apart from regulating cerebral blood flow, PGE2 also modulates BBB integrity and cell coupling. A recent study showed PGE2 could break down pericyteendothelial interaction by downregulating pericyte N cadherin and Cx43 in an EP1- and EP4-dependent manner (Perrot et al., 2020). PGE2 induces vascular inflammation via EP1 by activating NLRP3/Caspase1/IL-1β pathway, which boosts more PGE2 production by activating microsomal prostaglandin E synthase-1 (Perrot et al., 2018). EP4 activation induces endothelial migration via the cAMP/protein kinase A pathway (Khan et al., 2019). In addition, Aβ-evoked ET-1 release activated pericyte endothelin-A receptor (ET-A) causing capillary constriction and pericyte death (Nortley et al., 2019). Although these are not direct effects on BECs, the reduction in local blood flow induces ischemia thus negatively impacts on BBB integrity. Increased release of ET-1 is reported in AD astrocytes (Viejo et al., 2022). Increased astrocytic ET-1 could also impair BBB integrity. A previous study has reported the contributive role of ET-1 on BBB dissociation via increasing the production of endothelial MMP-2, 9, and VEGF. In addition, ET-1 could also reduce astrocytic AQP4, which might affect contact between endothelial and astrocytic endfeet, as well as water distribution (Hostenbach et al., 2016). Vascular inflammation can be induced by ET-1 through endothelial ET-A and ET-B. Activation of NF-κB is involved in ET-1-induced vascular inflammation, which includes increased PGE2 production via Ca2+uprise and COX2 activation (Lin et al., 2013, 2014), and immune cell migration (Montezano et al., 2007) (upregulated ICAM-1, vascular cell adhesion molecule 1 (Montezano et al., 2007), monocyte chemoattractant protein 1 (Chen et al., 2001), and E-selectin (Michinaga and Koyama, 2017; D’Orléans-Juste et al., 2019).
Matrix metalloproteinases
MMPs function as remodeling endopeptidases that degrade the extracellular matrix, including collagen, fibronectin, laminin as well as TJ-related proteins. Increased productions of MMP2 and MMP9 from reactive astrocytes stimulated by Aβ have been observed (Cabral-Pacheco et al., 2020). MMPs shift all the cells at BBB into proinflammatory states. Astrocytic MMP2 and MMP9 could in turn stimulate NF-κB activation leading to astrocytic chemokine expressions (Song et al., 2015). A previous study investigating the relationship between AD astrocytic ApoE and pericytes showed that proinflammatory pathway cyclophilin (CypA)/NF-κB/MMP9 was activated in pericytes leading to BBB breakdown (Bell et al., 2012). In endothelial cells, activations of NF-κB, cyclooxygenases (COX) and STAT3 also boost endothelial MMPs expression (Figure 4C).
ApoE4
Most of the ApoE protein in the CNS is synthesized by astrocytes. The geneAPOEε4(APOE4), one of the three alleles (ε2, ε3, and ε4), is associated with increased AD risk. ApoE protein is a high-affinity ligand for LRP1 and lowdensity lipoprotein receptor expressed on BECs, which are transmembrane receptors involved in receptor-mediated endocytosis of brain-derived molecules such as Aβ for efflux. ApoE is essential for BBB integrity by interacting with LRP1 on pericytes to block the CypA/NF-κB/MMP9 pathway (Bell et al. 2012). ApoE4, the protein product of theAPOE4allele, negatively affects LRP- and low-density lipoprotein receptor-dependent clearance and activates CypA / NF-κB /MMP9 pathway in pericyte, resulting in increased MMP9 release (Nikolakopoulou et al., 2021), and human-carryingAPOE4is more prone to BBB breakdown. The roles of astrocytic MMPs are depicted in Figure 4C.
NO and ROS
NO and ROS are commonly observed when astrocytes are stimulated. NO overproduction in reactive astrocytes is due to the upregulation of inducible nitric oxide synthase. NO exerts disruptive effects on BBB and downregulates endothelial TJ proteins. The probable underlying mechanism might involve the activation of cyclic guanosine monophosphate and protein kinase G (Wang et al., 2021; You et al., 2022). ROS together with decreased antioxidative capacity led to arachidonic acids-mediated inflammation pathology. Vascular inflammation is manifested as MMP activation and increased inflammatory mediators (Pun et al., 2009). ROS induces phosphorylation, redistribution, downregulation, and degradation of TJ-related proteins and cytoskeleton reorganization. The underlying mechanisms might include ROS-induced activation of the ROCK /MLC /MLCK pathway (Li et al., 2018). ROS also activates NRF2 /NAD(P)H dehydrogenase (NQO1) (antioxidative) pathway, possibly by a feedback mechanism (Alvarez et al., 2013). Figure 4A summarizes the signaling pathways activated by astrocytic NO and ROS in brain endothelial cells.
VEGF-A
VEGF-A is usually acknowledged as a detrimental factor for BBB integrity in various diseases, such as epilepsy, stroke, Parkinson’s disease, and multiple sclerosis (Shim and Madsen, 2018). VEGF-A increase is also observed in the CSF (Sopova et al., 2014), plasma (Cho et al., 2017), and frontal and parahippocampal cortex (Thomas et al., 2015) of AD patients. Inhibiting VEGF-A signaling through intraperitoneal administration of mouse VEGF-A164 protein improved cerebral blood flow and reduced BBB permeability through upregulating occludin and downregulating eNOS in APP/PS1 mice (Ali et al., 2022). Previous studies have shown that Aβ-stimulated astrocytes secrete increased levels of VEGF-A which is capable of triggering endothelial activation and inducing expressions of adhesion molecules (ICAM-1, vascular cell adhesion molecule 1) and MMP9 (Spampinato et al., 2017). As we have mentioned above, VEGF-A production in astrocytes can also be induced by VEGF-A, S100B, and IL-1β. VEGF signals through VEGFR2 expressed on BECs to activate various downstream pathways, including PI3K/Akt/eNOS, phosphoinositide phospholipase Cγ/ PKC/ ERK, p38 and Src, resulting in hyperpermeability (Figure 4B). Activation of phospholipase Cγ and eNOS correlates to gene downregulation of TJ proteins (occludin and claudin-5)in vitroandin vivo(Argaw et al., 2009, 2012). eNOS (S1176) activation disrupts endothelial junctions via producing NO. On the one hand, NO is transformed into toxic peroxynitrite (ONOO-) by reacting with ROS, which dissociates TJ and AJ complex (Ding et al., 2014). On the other hand, NO activates RhoAinduced hyperpermeability and Src-dependent VE-cadherin phosphorylation and internalization (Di Lorenzo et al., 2013). PKC could also activate RhoAmediated hyperpermeability (Appunni et al., 2021). Activation of Src and focal adhesion kinase is correlative to phosphorylation and internalization of the VE-Cadherin complex causing AJ disruption (Chen et al., 2012). MLCKinduced scaffold remodeling is an important event in VEGF-A-induced hyperpermeability, which can be triggered by upstream signal proteins including PKC, ERK, NO, and Ca2+(Shen et al., 2010). Astrocytic VEGF-A also acts as an autocrine to activate proinflammatory NF-κB in astrocytes (Ding et al., 2021). However, contradicting findings about the protective effect of VEGF are also reported. Coexpression of ANG1 and VEGF exhibits a promotive role for BBB integrity, but not VEGF alone in permanent distal middle cerebral artery occlusion mouse model (Shen et al., 2011). Administration of VEGF microcapsule (Spuch et al., 2010), nanospheres (Herrán et al., 2013; Herran et al., 2015), or mesenchymal stem cells that overexpress VEGF (Garcia et al., 2014) alleviate cognitive impairment, neurovascular repair and Aβ clearance in APP/PS1 models. Our previous study also observed suppressed VEGFR2 signaling in BECs treated with medium from Aβ-stimulated astrocyte (Yue et al., 2022a; Figure 4B), suggesting dysfunctional VEGF signaling in endothelial cells, which could be caused by secreted factors from Aβ-stimulated reactive astrocytes might contribute to BBB breakdown in AD brain.
Beneficial factors
A series of BBB promotive growth factors (GDNF, bFGF (Viejo et al., 2022), ANG1, TGFβ (Viejo et al., 2022), SHH, angiotensin-converting enzyme (Smit et al., 2021)), as well as RA, are upregulated in AD astrocytes surrounding Aβ or tau (Ji et al., 2008; Cheslow and Alvarez, 2016; Xing et al., 2019), suggesting defensive mechanisms of reactive astrocytes trying to restore BBB microenvironment. On the other hand, reactive astrocytes reduced secretion of brain-derived neurotrophic factor (Wang et al., 2019b), which might promote BECs function and inhibit vascular inflammation and oxidative stress via activating tyrosine receptor kinase B-mediated KLF2/hexokinase-1 pathway (Jin et al., 2021).
Conclusion and Perspective
In this review, we have summarized current studies reported on the distinctive characteristics of AD-induced astrocytes, covering findings from AD patients, AD-related transgenic animals, and astrocyte cellular models. Consistently, astrocytes in these disease models were all skewed into proinflammatory and oxidative profiles, as well as decreased glucose metabolism and glutamate reuptake. However, phagocytic activity for Aβ is overall increased, although some functional proteins were decreased. In AD patients and AD mouse models, astrocyte morphology is displayed in a probable district-dependent manner. Atrophic astrocytes with decreased cell somata and process number were widely observed in brain districts such as the entorhinal cortex and prefrontal cortex (Verkhratsky et al., 2019). However, hypertrophic astrocytes (also called reactive astrocytes) with increased cell bodies and possess thickness, as well as elongated processes (Zhou et al., 2019) mainly located around Aβ plaques in the hippocampus (Verkhratsky et al., 2016), along with increased distribution of AQP4 and Cx43, which might indicate astrocyte hypertrophy and redistribution of endfeet proteins facilitate astrocytesmediated Aβ clearance. Atrophic astrocyte morphology as well as swollen astrocytic endfeet could induce structural disruption of BBB. Swollen endfeet are observed in hypertrophic astrocytes around Aβ plaques (Bugiani et al., 2022), which might indicate that both atrophic and hypertrophic astrocytes in the AD brain contribute to BBB disruption.
A profile of proinflammatory and oxidative secretions released from astrocytes has been widely reported in different models. Proinflammatory mediators have been identified as playing a beneficial role at the beginning of inflammation to remove unwanted stimulants such as bacteria and virus (Soung and Klein, 2018), and become pathogenic as chronic proinflammatory states occur. The increased astrocytic secretions of cytokines and chemokines lead to the initial opening of BBB to allow infiltrating immune cells to the site of insults for debris clearance. In AD postmortem brain tissue, increased CD4+and CD8+T cells were observed near neurite plaques and microglia (Togo et al., 2002). Most studies just observed the detrimental role of T cells in activating microglial inflammation and neuronal death (Zieneldien et al., 2022). Only one study observed increased Aβ clearance and neurogenesis when administrating TH1 cells to APP/PS1 mice through CSF (Fisher et al., 2014). Prolonged cytokines and chemokines signaling will lead to BBB dysfunction. Astrocyte-derived secretions might contribute to BBB breakdown by affecting TJ, AJ expression and localization, endothelial inflammation, and immune cell infiltration.
Many studies have investigated the therapeutic potentials by targeting ADinduced astrocyte alterations (Table 2). Most pharmacological agents under interest focus on suppressing inflammatory responses and oxidative stress byinhibiting proinflammatory NLRP3, NF-κB, and STAT3 signaling, or enhancing antioxidant capacity such as increasing glutathione or SOD levels (Braidy et al., 2019; Jorda et al., 2020b). Inhibitors of S100β synthesis, such as arundic acid (ONO-2506), have been used for regulating astrocyte activation, showing some promising results in inhibiting cerebral infarction in stroke and AD. Others try to restore normal astrocytic metabolic ability (e.g. pantethine) (van Gijsel-Bonnello et al., 2017). In addition, ApoE-mimetic peptides have attracted attention aiming at restoring decreased astrocytic ApoE production or replacing deleterious ApoE4 in AD (Chernick et al., 2018). There is also a strategy focusing on inhibiting receptors that mediate neurotoxic activation of astrocytes, such as ET-B receptors which are highly expressed in reactive astrocytes. Another approach is to activate beneficial receptors such as estrogen receptors thus to suppress astrogliosis (reactive astrocytes) and increase the production of protective growth factors. Some interventions also aim to inhibit the excessive release of neurotransmitters (Smit et al., 2021). Pharmacological inhibition on reactive astrocytes cannot affect Aβ clearance, which might in turn aggravate Aβ pathology. This review discussed the relevance of astrocyte alterations in response to amyloid pathology in the context of BBB dysfunction. Maintaining astrocyte normal morphology and normal secretion profile seems to be the potential therapeutical strategy for BBB protection in AD. However, astrocyte pathology in AD is complicated and needs further investigation such as the functional differences between atrophic and hypertrophic astrocytes. In addition, more consolidated evidence is needed to reveal the causal relationship between BBB breakdown and biochemical secretions from AD astrocytes.

Table 2 |Current findings in potential compounds in modifying astrocyte pathology in AD models
Author contributions:The authors were responsible for all literature searches, data collection, writing, and submission of the manuscript, and approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Data availability statement:All relevant data are within the paper and its Additional files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional file:
Additional file 1:Full names of all abbreviations for Figures 1–4.

- 中国神经再生研究(英文版)的其它文章
- Mesenchymal stem cells, extracellular vesicles, and transcranial magnetic stimulation for ferroptosis after spinal cord injury
- Inducing prion protein shedding as a neuroprotective and regenerative approach in pathological conditions of the brain: from theory to facts
- Use of mesenchymal stem cell therapy in COVID-19 related strokes
- Brain organoids are new tool for drug screening of neurological diseases
- External anal sphincter electromyography in multiple system atrophy: implications for diagnosis, clinical correlations, and novel insights into prognosis
- New insights into the biological roles of immune cells in neural stem cells in post-traumatic injury of the central nervous system