
Mechanisms and treatment strategies of demyelinating and dysmyelinating Charcot-Marie-Tooth disease
2023-02-13 12:41NadgeHertzogClaireJacob
中国神经再生研究(英文版) 2023年9期
Nadège Hertzog, Claire Jacob
Abstract Schwann cells, the myelinating glia of the peripheral nervous system, wrap axons multiple times to build their myelin sheath. Myelin is of paramount importance for axonal integrity and fast axon potential propagation. However, myelin is lacking or dysfunctional in several neuropathies including demyelinating and dysmyelinating Charcot-Marie-Tooth disease. Charcot-Marie-Tooth disease represents the most prevalent inherited neuropathy in humans and is classified either as axonal, demyelinating or dysmyelinating, or as intermediate. The demyelinating or dysmyelinating forms of Charcot-Marie-Tooth disease constitute the majority of the disease cases and are most frequently due to mutations in the three following myelin genes: peripheral myelin protein 22, myelin protein zero and gap junction beta 1 (coding for Connexin 32) causing Charcot-Marie-Tooth disease type 1A, Charcot-Marie-Tooth disease type 1B, and X-linked Charcot-Marie-Tooth disease type 1, respectively. The resulting perturbation of myelin structure and function leads to axonal demyelination or dysmyelination and causes severe disabilities in affected patients. No treatment to cure or slow down the disease progression is currently available on the market, however, scientific discoveries led to a better understanding of the pathomechanisms of the disease and to potential treatment strategies. In this review, we describe the features and molecular mechanisms of the three main demyelinating or dysmyelinating forms of Charcot-Marie-Tooth disease, the rodent models used in research, and the emerging therapeutic approaches to cure or counteract the progression of the disease.
Key Words: Charcot-Marie-Tooth disease; rodent models; emerging treatments; demyelination and dysmyelination; endoplasmic reticulum stress; gene therapy; myelin; repair; Schwann cells; unfolded protein response
Introduction
Charcot-Marie-Tooth disease (CMT) is the most common form of inherited peripheral neuropathy with a prevalence of 1 individual in 2500 (Fridman and Saporta, 2021). Disease onset can occur at different periods of life ranging from infancy to late adulthood, but symptoms mostly appear between 5 and 25 years of age. Most patients present high-arched feet also known as pes cavus deformity, weak ankle dorsiflexion, decreased or absent deep tendon reflexes, weakness and atrophy of distal limb muscles, and mild to moderate distal sensory loss. Certain CMT subtypes can also be accompanied by hearing loss, optic neuropathy, or other central nervous system dysfunctions (Stavrou et al., 2021a). CMT progression is slow and disabilities accumulate gradually over time. CMT is classified according to the inheritance pattern (autosomal dominant, autosomal recessive, and X-linked), to motor nerve conduction velocity, and to the type of primary defect (primarily demyelinating or dysmyelinating, primarily axonal or intermediate types), with CMT1 and CMT4 regrouping the primarily demyelinating and dysmyelinating forms, CMT2 the primarily axonal forms and I-CMT the intermediate forms (Pisciotta et al., 2021). Furthermore, the genetic diversity of CMT results in several subtypes with distinguished phenotypic features. Indeed, over 120 causative genes have been identified and many of them present several variants (Juneja et al., 2019). Consequently, the pathomechanisms involved in the different types of CMT are diverse. They can affect the compact and non-compact myelin, cell signaling, transcription factors, mitochondria, cytoskeleton, transporters, channels, protein quality control machinery as well as axonal transport (Stavrou et al., 2021b). However, 90% of CMT patients suffer from a demyelinating or dysmyelinating form of CMT characterized by a motor nerve conduction velocity below 35 m/s in the upper limb, the most common forms being CMT type 1A (CMT1A), CMT type 1B (CMT1B), and X-linked CMT type 1 (CMT1X), which are caused by mutations in the genes coding for peripheral myelin protein 22 (PMP22), myelin protein zero (P0) and connexin 32 (Cx32), respectively. In the course of these demyelinating or dysmyelinating CMTs, Schwann cells (SCs) do not fully differentiate, dedifferentiate or produce unstable myelin. Therefore, myelin is either not formed or deteriorates, axons demyelinate and finally degenerate. To date, there is neither cure nor effective treatment available on the market to impede disease progression.
Search Strategy and Selection Criteria
The search strategy and selection criteria were limited to articles published in peer-reviewed journals. A literature review of articles was conducted by searching in the National Library of Medicine (PubMed) database and was updated until September 2022, with the following key words and combinations of them: Charcot-Marie-Tooth disease, CMT1A, CMT1B, CMT1X, PMP22, MPZ, Cx32, rodent models, and therapy. No limit was placed on the year of publication or authorship. Relevant recent articles or reviews were used for each section and further selection was done by reading the title, abstract and full article. The list of references for each study was also screened to identify other useful studies. Older publications were cited to give credit to the original authors when necessary.
Demyelinating and Dysmyelinating Forms of Charcot-Marie-Tooth Disease and Rodent Models
In this section, we will introduce the most frequent forms of CMT, namely CMT1A, CMT1B, and CMT1X, and the associated rodent models. The diverse CMT subtypes, their molecular pathomechanisms, clinical features, and rodent models have been reviewed in detail elsewhere (Niemann et al., 2006; Bosco et al., 2021; Fridman and Saporta, 2021; Stavrou et al., 2021b).
CMT1A
CMT1A is a demyelinating and dysmyelinating and the most frequent form of CMT, present in 50% of patients. CMT1A also accounts for 70–80% of all CMT1 (Szigeti et al., 2006). At the neuropathological level, CMT1A is characterized by abnormal myelination, onion bulb formation, and decreased density of myelinated fibers, which leads to axonal degeneration and the clinical features mentioned above (reviewed in van Paassen et al., 2014). CMT1A has been considered as primary demyelinating, caused by repetitive demyelination and remyelination. Indeed, the presence of onion bulbs detected in the nerve biopsies is typical of recurrent de- and remyelination and occurs when multiple SCs wrap several times around one axon but fail to produce compact myelin (Pleasure and Towfighi, 1972; Tracy et al., 2019). However, recent findings from electrophysiological data of CMT1A patients suggest that CMT1A may be primarily dysmyelinating rather than demyelinating and onion bulb formation would be the consequence of an abnormal SC organization around axons during development (reviewed in Li, 2017). Indeed, nerve conduction velocity is uniformly slowed and remarkably consistent among CMT1A patients, which seems incompatible with repetitive de- and remyelination events that would rather lead to highly variable nerve conduction velocity values among patients. CMT1A results from a 1.4 Mb tandem duplication containing thePMP22gene on chromosome 17p11.2, leading to PMP22 overexpression (Hoogendijk et al., 1991; Patel et al., 1992; Pentao et al., 1992; reviewed in Suter and Scherer, 2003). ThePMP22gene codes for a 22-kDa tetraspan hydrophobic glycoprotein, which represents 2–5% of peripheral nerve myelin proteins and can on its own induce the myelin-like assembly of lipid bilayers (Snipes et al., 1992; Mittendorf et al., 2017; Marinko et al., 2021).
Several CMT1A rodent models have been generated (Huxley et al., 1996, 1998; Magyar et al., 1996; Sereda et al., 1996; Verhamme et al., 2011). The transgenic mouse lines C22 and C61 are two widely used CMT1A mouse models. C22 heterozygous mice carry seven copies of humanPMP22transgene and develop a progressive peripheral neuropathy with demyelination and remyelination of medium-to-large caliber axons, as well as onion bulbs, resembling the human pathology observed in CMT1A (Huxley et al., 1996, 1998; Kinter et al., 2013). In contrast, C61 heterozygous mice, which carry four copies of humanPMP22transgene, develop only mild demyelination and no visible phenotype; however, homozygotes of this line, carrying eight copies, develop a peripheral neuropathy similar to C22 heterozygous mice (Huxley et al., 1998). In these models, the demyelination phenotype is more severe than in CMT1A patients and the disease onset occurs earlier (Huxley et al., 1996, 1998). The C3-PMP mouse line is another CMT1A mouse model. This line spontaneously arose after backcrossing the C22 mouse line to C57BL/6j mice for more than 10 generations and was originally described to carry three to four copies of the humanPMP22transgene (Verhamme et al., 2011), but more recent work showed by using digital droplet PCR that this line carries actually five copies of the transgene (Prior et al., 2022). C3-PMP mice present a moderate-to-severe phenotype with a milder neuromuscular impairment as C22 mice, a lower degree of axon dysmyelination, and a stable number of myelinated fibers over time (Verhamme et al., 2011; Prior et al., 2022). Lastly, thePMP22transgenic mouse line carrying between sixteen and thirty copies of the humanPMP22gene shows very severe hypomyelinating neuropathy, with almost a complete lack of myelin. In this line, SC differentiation is strongly impaired, leading to continuous SC proliferation but without onion bulb formation (Magyar et al., 1996). Despite the close phenotypic traits in common with the clinical representation of CMT1A, more copies are needed in the mouse to recapitulate the phenotype of thePMP22duplication occurring in humans. This is because the humanPMP22transgene is expressed at a lower level as compared to the murinePmp22gene (Huxley et al., 1998). In addition, the genomic and epigenomic consequences caused byPMP22duplication in humans cannot be recapitulated (Fridman and Saporta, 2021). Another well-known CMT1A rodent model is thePMP22transgenic rat, which carries three copies of a 43 kb restriction fragment containing thePMP22transcription unit as well as 7 kb upstream of exon 1A and 4 kb downstream of exon 5. This model presents many morphological (e.g., demyelination and onion bulb formation), electrophysiological and behavioral features of human patients suffering from CMT1A (Sereda et al., 1996).
PMP22 is necessary for myelinogenesis, appropriate myelin thickness and stability, and thus for proper development and maintenance of myelinated axons in the peripheral nervous system (PNS). The increased amount of PMP22 in CMT1A results in the destabilization of the myelin sheath structure, demyelination, and finally secondary axonal loss. Several studies identified an important role for PMP22 in the organization and localization of myelin lipids as well as lipid metabolism in SCs (Lee et al, 2014; Fledrich et al., 2018; Zhou et al., 2019). This is further highlighted by studies on CMT1A rats. Indeed, the CMT1A rat presents an altered transcription of genes implicated in myelin lipid biosynthesis, resulting in an imbalanced stoichiometry between myelin lipids and myelin proteins and thus a destabilization of the myelin structure and SC functions (Fledrich et al., 2018; Stavrou et al., 2021b). In healthy individuals, only a small proportion of PMP22 is targeted to the plasma membrane, while the majority of PMP22 is degraded directly after translation, due to inefficient folding (Pareek et al., 1997). Overexpression of PMP22 has been shown to affect its cellular localization. For instance, cholesterol and PMP22 are retained in lysosomes in the CMT1A rat (Zhou et al., 2020) and fibroblasts isolated from CMT1A patients and SCs derived from C22 mice show cytosolic PMP22 aggregates and a decreased proteasomal activity (Fortun et al., 2006; Lee et al., 2018). In healthy conditions, misfolded PMP22 is detected by the endoplasmic reticulum (ER) quality control and targeted to the proteasome for degradation (Fortun et al., 2006). The presence of PMP22 aggregates in CMT1A SCs suggests that the ER quality control system capacity is exceeded, leading to ER stress and activation of the unfolded protein response (UPR) (Marinko et al., 2020; Figure 1). In addition to interacting with and regulating factors involved in cholesterol biosynthesis (Zhou et al., 2019), PMP22 has other functions. For instance, PMP22 stabilizes α6β4 integrin and laminin complexes and acts as a bridge in the communication between SCs and the extracellular matrix (Amici et al., 2006). In addition, PMP22 interacts with P0 and promotes myelin compaction (D’Urso et al., 1999).In normal conditions, myelin proteins are functional and present in the proper stoichiometry, allowing myelin compaction and optimal axon-SC communication. Axonderived NRG1-type III can bind the ErbB2/3 receptor on SCs and repressesNrg1-type Iexpression via MEK/ERK signaling. Myelin protein gene transcription is maintained by Krox20, while transcription of negative regulators of myelination (c-Jun, Id2, and Sox2) is silenced, and the activity of PI3K/AKT and MEK/ERK signaling pathways is balanced, thereby maintaining SCs in their differentiation state. In the ER, the misfolded myelin proteins are normally degraded via the ERAD and proteasome pathway. In the case of CMT, the presence of non-functional/dysfunctional myelin proteins and/or the reduced or increased levels of functional myelin proteins perturbate the myelin stoichiometry and impair axon-SC communication. This results in axon dysmyelination, hypomyelination, and demyelination. In CMT1A (1), the perturbed axon-glia communication decreases PI3K/AKT signaling activity, which results in MEK/ERK hyperactivation. The imbalanced activity of PI3K/AKT and MEK/ERK signaling pathways activates the transcription of SC immature and repair markers, leading to impaired SC differentiation and myelination. The absence of axon-SC contact prevents NRG1-type III binding to ErbB2/3 receptors, leading toNrg1-type Iexpression. SC-derived NRG1-type I is secreted in an autoparacine way and activates ErbB2/3 receptors, which signal through MEK/ERK and further trigger the expression of negative regulators of myelination. The chronic NRG1-type I expression also leads to hypermyelination and onion bulb formation. In CMT1B (2) and CMT1X (3), misfolded mutant P0 and Cx32 aggregate and accumulate in the ER. Furthermore, some mutant misfolded forms of P0 and Cx32 can trap the wild-type forms of P0 and Cx32 in the ER. Consequently, the levels of functional P0 and Cx32 are reduced in the myelin sheath. PMP22 aggregates are also observed in the ER, cytoplasm, and lysosomes in CMT1A. The accumulation of protein aggregates overloads the ERAD capacity, leading to ER stress and activation of the three UPR arms. The PERK-mediated CHOP activation and the cleaved form of ATF6 reduce myelin gene transcription. GADD34 is then activated by CHOP and dephosphorylates eIF2A. The dephosphorylated form of eIF2A restores normal translational levels and activates MER/ERK. How eIF2A induces MEK/ERK activation is still unknown. In parallel, the IRE1 arm of the UPR may upregulate c-Jun levels via c-Jun N-terminal kinase, leading to Krox20 downregulation and reduced myelin gene transcription. The decreased levels of myelin proteins may then reduce ERAD overloading and ER stress. In CMT1A, CMT1B, and CMT1X, MEK/ERK-mediated MCP-1/CCL2 expression in SCs recruits and activates macrophages, leading to axonal damage and demyelination. In CMT1X, endoneurial fibroblasts form cell-cell contact with macrophages and secrete the macrophage activator CSF-1. The resulting inflammation further impairs SC differentiation. How CMT1X SCs induce CSF-1 expression in fibroblasts is still unknown. ATF4: Activating transcription factor 4; ATF6: activating transcription factor 6; CHOP: CCAAT/enhancer-binding protein homologous gene;c-Jun: gene coding for Jun proto-oncogene; CMT1A: Charcot-Marie-Tooth disease type 1A; CMT1B: Charcot-Marie-Tooth disease type 1B; CMT1X: X-linked Charcot-Marie-Tooth disease type 1; CSF-1: colony-stimulating factor 1; Cx32: connexin 32; eIF2A: eukaryotic translation initiation factor 2A; ERAD: endoplasmic reticulum-associated degradation; ErbB2/3: heterodimer of erb-b2 receptor tyrosine kinases 2 and 3; GADD34: DNA damage inducible protein 34;Id2: gene coding for inhibitor of DNA binding 2; IRE1: inositol-requiring enzyme 1; JNK: c-Jun N-terminal kinase; Krox20: also called Egr2, early growth response 2; MCP-1/CCL2: monocyte chemoattractant protein 1/C-C motif chemokine ligand 2; MEK/ERK: extracellular signal-regulated kinase/mitogen-activated protein kinase;Nrg1-type I: gene coding for neuregulin 1 type I; NRG1-type I: neuregulin 1 type I; NRG1-type III: neuregulin 1 type III; P0: myelin protein zero; PERK: protein kinase RNA-like endoplasmic reticulum kinase; PI3K/AKT: phosphoinositide-3 kinase/v-Akt murine thymoma viral oncogene homolog 1; PMP22: peripheral myelin protein 22;Sox2: gene coding for SRYbox transcription factor 2; UPR: unfolded protein response; XBP1: X-box-binding protein 1.
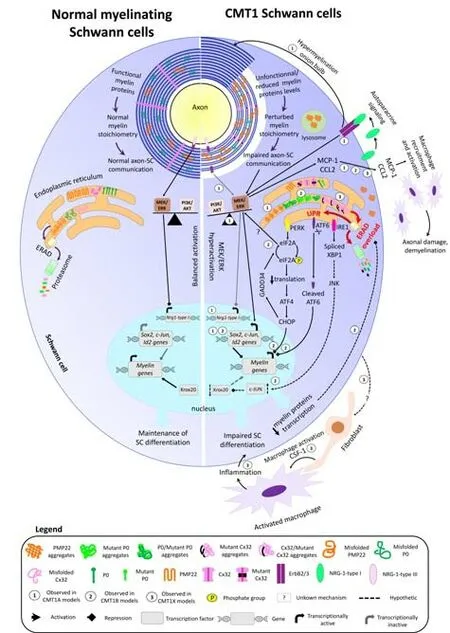
Figure 1|Overview of myelin structure, molecular pathways, and gene transcription in normal myelinating SCs (left) and in demyelinating/dysmyelinating CMT SCs (right).
CMT1B
CMT1B, which accounts for 10% of the demyelinating or dysmyelinating forms of CMT, is caused by mutations affecting theMPZgene coding for P0. CMT1B is usually classified into two categories based on the age of onset, and clinical and pathological features. The early-onset form is characterized by severe demyelination and disability in childhood together with a decrease in disease progression during adulthood (Shy et al, 2004; Bai et al., 2006). In contrast, the adult forms are mostly axonal neuropathies, characterized by a rapid progression but with very mild demyelination (Sanmaneechai et al., 2015; Howard et al., 2021). P0 represents around 50% of myelin proteins in the PNS (Siems et al., 2020) and is specifically expressed in the SC lineage. P0 plays an essential function in myelin formation and maintenance and is also required to maintain nodal and paranodal integrity (Brügger et al., 2015; Bolino, 2021). P0 consists of an extracellular immunoglobulin-like domain, a transmembrane domain, and an intracellular domain. Myelin compaction occurs when extracellular P0 domains on opposing myelin wraps are brought in contact and form a homotetramer, thus reducing the intermembrane distance (Raasakka and Kursula, 2020).
To understand the physiopathology of the disease, several CMT1B rodent models have been generated. One broadly used model is theMpzR98Cknockin mouse. This line has been used in its heterozygous form (MpzR98C/+) and its homozygous form (MpzR98C/R98C). TheMpzR98Cline has been generated by inserting the R98C mutation found in human CMT1B, characterized by severe and early dysmyelination and demyelination. BothMpzR98C/+andMpzR98C/R98Cmice show the same pathological features of CMT1B comprising hypomyelination, weakness at early age and slow nerve conduction velocities, reduced number of myelinated axons and segmental demyelination. However, while SC development is delayed in heterozygous animals, SCs of the homozygous animals are arrested at the promyelinating stage and animals display a more severe phenotype (Saporta et al., 2012). Another well-known CMT1B rodent model is the transgenic mouse lineMpzS63del, in which theMpzS63deltransgene was inserted randomly. In humans, this mutation leads to a milder form of CMT1B with late-onset demyelination and thinner myelin.MpzS63delnerves show hypomyelination and demyelination as well as onion bulb formation and the animals recapitulate the electrophysiological features of CMT1B patients as well as clinical progression with age. However, axonal loss is not observed (Wrabetz et al., 2006). Both CMT1B rodent models show retention of the mutant protein in the ER and subsequent activation of the UPR (Wrabetz et al., 2006; Saporta et al., 2012).
Recently, Veneri et al. (2022) generated a new CMT1B mouse model, theMpzD61N/+line, which contains an additional glycosylation site on P0 extracellular sequence. This line develops a severe form of CMT1B early after birth, which worsens over time and is characterized by a severe loss of motor function. At the morphological level, axons are dysmyelinated, demyelinated and myelin compaction is impaired. Interestingly, P0D61N/+proteins do not form aggregates in the ER. Instead, they are normally targeted to the plasma membrane. This is in contrast with P0S63deland P0R98Cmutant proteins, which never reach myelin (Wrabetz et al., 2006; Saporta et al., 2012). In the myelin sheath, P0D61Nprotein acts in a dominant-negative fashion. A potential mechanistic hypothesis could be that the gained glycosylation on P0 extracellular domain impedes the formation of P0 tetramers and thus impairs myelin compaction, but this remains to be demonstrated (Veneri et al., 2022).During its trafficking through the Golgi to the plasma membrane, P0 is glycosylated on its extracellular domain. This post-translational modification is essential for proper P0-P0 homophilic interactions and thus myelin compaction (Griffith et al., 1992). However, as mentioned above, the addition of glycosylation sites on the extracellular domain of P0 results in impaired homophilic interactions and myelin decompaction (Veneri et al., 2022). The intracellular domain of P0 is also required for efficient myelin compaction. Indeed, through signal transduction, the cytoplasmic domain interacts with the cytoskeleton and mediates myelin adhesion (Xu et al., 2001; Gaboreanu et al., 2007). Moreover, the intracellular domain is also required for proper P0 trafficking to the myelin plasma membrane (Fratta et al., 2019). CMT1B is associated with a wide range of mutations affecting each of the three P0 domains. While some P0 mutant proteins are retained in the ER and trigger UPR, other mutants are targeted to the myelin and non-myelin plasma membranes, interfere with the function of wild-type P0 and impair myelin adhesion (Wrabetz et al., 2006; Bai et al., 2018; Veneri et al., 2022; Figure 1).
CMT1X
CMT1X is an X-linked dominant form of CMT and accounts for 5–10% of all CMT cases. It is caused by a mutation of theGJB1gene coding for Cx32. As expected for X-linked diseases, men are more severely affected than women, and disease onset occurs already during childhood (Shy et al., 2007). In contrast to the other forms of demyelinating CMT, CMT1X is considered a mixed axonal and demyelinating pathology, and axonal loss can occur already in childhood. Patients can also present encephalopathy syndromes under metabolic stress conditions (Tian et al., 2021). Cx32 is principally localized at the paranodes and Schmitt-Lanterman incisures, where it forms hexameric structures called connexons or hemichannels. Two hemichannels interact to form a gap junction (reviewed in Cisterna et al., 2019). By forming a channel for ions and signaling molecules between the myelin sheath and axons, Cx32 gap junctions are essential for the functional integrity of axons. Moreover, Cx32 hemichannels may also impact the myelination process. Indeed, electrical stimulation triggers axonal adenosine triphosphate release which activates P2Y purinoreceptors on the SC membrane. Subsequently, inositol triphosphate activation mediates Ca2+increase in the SC cytosol and mitochondrial matrix. The suppression of this pathway has been shown to trigger hypomyelination. The increased Ca2+levels in SCs may trigger Cx32 hemichannel opening and the release of adenosine triphosphate and Ca2+, further propagating Ca2+signaling (reviewed in Bortolozzi, 2018 and Qiu et al., 2022). More than 450 nonsense, frameshift, missense mutations, deletions, and insertions have been associated with CMT1X, mostly resulting in loss of function phenotypes. Some of the mutant Cx32 proteins are either not synthesized, or do not reach the plasma membrane and are retained in the ER and Golgi apparatus. Other mutant Cx32 proteins are inserted into the plasma membrane but do not form functional gap junctions or form gap junctions with altered properties as compared to wild-type channels (Bortolozzi, 2018; Qiu et al., 2022; Figure 1).
The main rodent model used so far is theGjb1-null mouse. These mice develop peripheral neuropathy at the age of 3 months and show demyelinated and remyelinated axons. Interestingly, motor axons are more affected than sensory axons. Histopathological features as well as disease progression are very close to the human CMT1X (Suter and Scherer, 2003). However, this model cannot be used to test drugs attempting to restore the biochemical properties of mutated Cx32, and only 10% ofGjb1mutations are null alleles (Mones et al., 2012). Therefore, several knock-in lines with variousGjb1mutations have been generated. Loss of function mutations affect either cell trafficking of Cx32 or the function of the hemichannel. In the first category, mutated Cx32 is retained in the ER and is not present in the myelin sheath. In the second one, mutated Cx32 proteins are assembled into dysfunctional connexons (Mones et al., 2012). Mones et al. (2012) generated two transgenic lines, namelyGjb1G12S, in which Cx32 cell trafficking is affected, andGjb1S26L, in which the connexon is dysfunctional. In addition, some Cx32 mutants have dominant-negative interactions. For instance, theCx32R142Wmutant localizes into the Golgi, reduces the level of endogenous Cx32, and triggers demyelination. In addition, this mutant also causes reversible central nervous system dysfunctions, a feature which is not observed with loss of function mutations or inGjb1-null mice (Jeng et al., 2006). Molecular Mechanisms of Demyelinating and Dysmyelinating Charcot-Marie-Tooth Disease Demyelinating or dysmyelinating CMTs can result from defects in various biological processes in SCs including myelin compaction, exchanges across myelin, the link between the extracellular matrix and the cytoskeleton, transcription of myelin genes, regulation of endosomal pathways, receptor endocytosis and recycling, trafficking, degradation, mitochondrial dynamics. The underlying molecular mechanisms are thus diverse and depend on the function of the affected gene. These mechanisms can be due to loss of function mutations or to a toxic gain of function. In the case of CMT1A, CMT1B, and CMT1X, mutant proteins or an excessive number of wild-type proteins can be targeted to myelin but lead to dysfunctional or unstable myelin, which can eventually result in demyelination, or can accumulate in the ER and form protein aggregates, which induces cytotoxicity, or can affect protein biosynthesis, processing or degradation and deregulate signaling pathways, leading to misregulation of gene expression and thereby to the maintenance of a non-myelinating SC phenotype (Figure 1). In this section, we have detailed the molecular mechanisms that are the best described for CMT1A, CMT1B, and CMT1X. It is however important to note that the physiopathology of these CMTs and other CMTs remains partially understood and a lot more work is needed to fully characterize the molecular mechanisms responsible for the different CMT subtypes.
CMT1 Schwann cell phenotype shows similarities with the repair Schwann cell phenotype
At first glance, demyelinating and dysmyelinating neuropathies and Wallerian degeneration (WD) appear to be two distinct events or conditions. However, they share some similarities. Indeed, the inflammation process mediated by immune cells, the mediators of SC demyelination as well as the molecular pathways implicated in axonal degeneration have common molecular features in WD and CMT1 (Martini et al., 2013). Although WD is necessary for peripheral nerve regeneration, a chronic impairment of SC differentiation has a deleterious effect on axonal integrity and nerve function. Indeed, when CMT1A rats are crossed with Wlds transgenic rats, in which WD is slowed down, axonal degeneration is delayed (Meyer zu Horste et al., 2011). Both repair SCs and SCs of CMT rodent models express the transcription factor c-Jun (Jun proto-oncogene), the most important factor triggering SC conversion into the repair SC phenotype (Arthur-Farraj et al., 2012; D’Antonio et al., 2013; Fledrich et al., 2014). C-Jun can act downstream of the phosphorylated extracellular signal-regulated kinase and of mitogen-activated protein kinase (MEK/ERK) signaling and is involved in SC demyelination after lesion. Indeed, c-Jun reduces myelin gene expression and blocks SC differentiation (Harrisingh et al., 2004; Arthur-Farraj et al., 2012; Napoli et al., 2012; Fazal et al., 2017). Furthermore, c-Jun has been found to be upregulated in patients affected by peripheral neuropathies (Hutton et al., 2011). This may indicate that SCs are maintained in an immature state. Interestingly, SC differentiation in the CMT1A rat is defective and these SCs express high levels of immature and injury markers throughout life, together with low levels of myelin proteins. In parallel, a strong reduction of phosphoinositide-3 kinase/v-Akt murine thymoma viral oncogene homolog 1 (PI3K/AKT) signaling leading to hyperactivation of the MEK/ERK pathway occurs and contributes to the differentiation defect (Fledrich et al., 2014). The resulting imbalance between PI3K/AKT and MEK/ERK signaling may be the consequence of insufficient neuregulin 1 (NRG1) signaling in SCs of CMT1A rats (Figure 1). Indeed, NRG1 activates PI3K/AKT signaling and treatment of CMT1A rats early after birth with NRG1 restores the balance between PI3K/AKT and MEK/ERK signaling and favors SC differentiation and myelination (Fledrich et al., 2014). NRG1 belongs to the epidermal growth factor-like factors and binds to the ErbB2/3 tyrosine kinase receptors expressed by SCs. NRG1 exists in different isoforms, including NRG1-type I and NRG1-type III, which are both expressed in the PNS (Nave and Salzer, 2006). During development, axonal NRG1-type III modulates the activation of promyelinating factors and thus myelin gene transcription. Axonal NRG1-type III is necessary for SC proliferation, differentiation and myelination, and NRG1-type III levels determine myelin thickness (Michailov et al., 2004; Nave and Salzer, 2006).
Surprisingly, NRG1-type I is secreted by SCs of CMT1A rodent models in an autoparacrine way throughout life and is responsible for disease progression by inducing MEK/ERK signaling and impeding SC differentiation. Indeed, chronic NRG1-type I expression results in axon hypermyelination and onion bulb formation, a typical morphological and pathologic feature of CMT1A (Figure 1). In addition, increased NRG1-type I expression was also observed in nerve biopsies of patients suffering from demyelinating neuropathies (Fledrich et al., 2019). While ablation of NRG1-type I in CMT1A rats worsens the disease phenotype early after birth, it normalizes the expression of immature and injury markers, promotes myelination, and ameliorates the disease phenotype in young adult and old CMT1A mice. This indicates that the supportive functions of SC-derived NRG1-type I in CMT1A mice are restricted to early postnatal stages (Fledrich et al., 2019). In adult mice, axonal NRG1-type III signals through MEK/ERK and represses NGR1-type 1 expression by SCs. Upon axonal loss, NRG1-type I is re-expressed by SCs during WD and is necessary for nerve repair and remyelination (Stassart et al., 2013). Therefore, the increased NRG1-type I expression in CMT1A SCs may be the consequence of impaired access to axonal NRG1-type III signaling during myelination, due to a decreased contact area with the axon (Fledrich et al., 2014, 2019).
Activation of the MEK/ERK pathway is also observed in mutant SCs of CMT1X models and triggers the expression of monocyte chemoattractant protein 1/ C-C motif chemokine ligand 2 (MCP-1/CCL2), which recruits and activates macrophages (Figure 1). Indeed, inhibition of ERK phosphorylation or knocking out MCP-1 expression inGjb1-null mice improved myelination, decreased axonal degeneration, and ameliorated the phenotype in a rodent model of CMT1X (Groh et al., 2010). MCP-1/CCL2-mediated macrophage recruitment and activation also take place in CMT1A and CMT1B mouse models and has also been associated with axonal demyelination and activation of MEK/ERK signaling pathway (Fischer et al., 2008; Kohl et al., 2010). SC demyelination inGjb1-null mice is also dependent on colonystimulating factor 1 (CSF-1)-mediated macrophage activation, which occurs independently of ERK signaling and may further impair SC differentiation and myelination (Figure 1). Indeed, knocking out CSF-1 inGjb1-null mice improved myelin preservation and prevented upregulation of immature and injury markers. Interestingly, endoneurial fibroblasts release CSF-1 inGjb1-null mice. HowGjb1-null SCs trigger CSF-1 release by endoneurial fibroblasts is however still unknown (Groh et al., 2015).
Endoplasmic reticulum-stress and unfolded protein response
Several studies focused on the UPR to explain the pathomechanisms of CMT1B (Saporta et al., 2012); however, UPR activation is also observed in CMT1X (reviewed in Bortolozzi, 2018) and in CMT1E, the latter resulting from point mutations in thePMP22gene that lead to changes in single AA (Marinko et al., 2020). Concerning CMT1A, increased expression of PMP22 protein due toPMP22gene duplication may lead to the accumulation of PMP22 in the ER, but this still needs to be demonstrated in the nerves of CMT1A patients (Marinko et al., 2020). In the rough ER, a specialized surveillance system, the ER-protein quality control, ensures that proteins are properly folded so that only functional proteins are delivered to the plasma membrane, whereas misfolded proteins are targeted to the ER-associated degradation (ERAD) and proteasome machineries to avoid the toxic accumulation of non-functional proteins (reviewed in Wiseman et al., 2022). The importance of ERAD in SC myelin integrity has recently been demonstrated (Volpi et al., 2019). In SCs, the myelin protein synthesis rate is high in normal conditions but only a small fraction of myelin proteins reaches the plasma membrane. In addition, the surface area of one myelinating SC is much larger than other mammalian cells and results in a high demand for protein and lipid synthesis. This renders SCs even more vulnerable to the accumulation of misfolded proteins (Lin and Stone, 2020). ER stress occurs when the ER-protein quality control capacity is overloaded due to the abnormal accumulation of misfolded proteins (Lin and Stone, 2020; Wu et al., 2021). In demyelinating or dysmyelinating forms of CMT, several Cx32, P0, and PMP22 mutant proteins are misfolded and thereby exhaust the ER-protein quality control capacity. In this situation, SCs activate the UPR through three ER-stress sensors: protein kinase RNAlike ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). Briefly, IRE1 fosters the activation of genes implicated in ERAD by promoting the splicing of X-box-binding protein 1 (XBP1) mRNA. Upon cleavage, ATF6 increases the levels of ER chaperone proteins to improve protein folding. On the other hand, activation of PERK leads to eukaryotic translation initiation factor 2A (eIF2A) phosphorylation and attenuation of general protein synthesis. In parallel, PERK increases the translation of activating transcription factor 4, which in turn induces the expression of CCAAT/enhancer-binding protein homologous gene (CHOP). CHOP upregulates DNA damage-inducible protein 34 (GADD34), which dephosphorylates eIF2A to restore normal translation rates. The activation of these three ER-stress transducers reduces myelin gene expression and can trigger SC demyelination and potentially death when the UPR is persistently activated (Sidoli et al., 2016; Volpi et al., 2017; Fridman and Saporta, 2021; Figure 1).
The two main mutations that result in the accumulation of misfolded proteins in the ER and in UPR activation areMpzS63delandMpzR98C. InMpzS63delmice, ablation ofGadd34rescues hypomyelination and demyelination and restores motor functions (Pennuto et al., 2008). Therefore, stabilizing the levels of phosphorylated eIF2A (p-eIF2A) maintains translation rates necessary for the preservation of myelin without overloading the ER with misfolded proteins (D’Antonio et al., 2013). Paradoxically, PERK ablation in SCs could partially restore myelination, independently of CHOP and despite the decreased p-eIF2A levels and P0 accumulation in the ER. This indicates that pathways other than the UPR may be perturbated by PERK and sheds light on another mechanism contributing to CMT1B pathogenesis inMpzS63delmice.
Interestingly, transcriptome analysis ofMpzS63delnerves shows induction of factors associated with SC injury and negative regulators of myelination including inhibitor of DNA binding 2 (Id2), c-Jun, and SRY-box transcription factor 2 (Sox2), together with downregulation of genes involved in lipid, sterol, and cholesterol biosynthesis. However, expression of the transcription factor Krox20, myelin basic protein, and PMP22 was surprisingly unaffected. Therefore, inMpzS63delmice, the synthesis of myelin proteins and myelin lipids is uncoordinated (D’Antonio et al., 2013). Moreover, ER-stress and UPR are not impacted inMpzS63delmice in which eIF2A is prevented from phosphorylation, but SCs show a more pronounced differentiation and myelination delay together with a stronger upregulation of c-Jun levels and a striking increase in phosphorylated extracellular signal-regulated kinase levels. Consequently, axon dysmyelination and demyelination inMpzS63delmice are more severe in the absence of p-eIF2A. This study showed that eIF2A phosphorylation has a protective role in CMT1B. How the inhibition of eIF2A phosphorylation triggers ERK activation remains however unclear (Scapin et al., 2020; Figure 1). While c-Jun, Sox2, and Id2 negatively affect SC myelination by blocking Krox20-driven expression of myelin genes (Parkinson et al., 2008; Roberts et al., 2017), their upregulation inMpzS63delmice has a protective function (Florio et al., 2018). Indeed, Sox2 and Id2 limit SC differentiation and prevent excessive accumulation of mutant P0 proteins and chronic ER stress (Figure 1). Furthermore, c-Jun upregulation also reduces the loss of sensory fibers and improves sensory-motor performances in the C3 mouse model (Hantke et al., 2014). A slight upregulation of c-Jun levels in SCs may thus protect from neurological damages. Indeed, while mice highly overexpressing c-Jun in SCs develop axonal hypomyelination, myelin is unaffected in mice that moderately overexpress c-Jun in SCs (Fazal et al., 2017). Normal myelination is thus compatible with a moderate increase of c-Jun levels, which is observed in SCs of CMT1A and CMT1X rodent models (Hantke et al., 2014; Klein et al., 2014; Fazal et al., 2017; Jessen and Mirksy, 2022). Of note, upregulation of negative regulators of myelination and c-Jun target genes is also observed in theMpzD61Nmouse but without upregulation of UPR markers (Veneri et al., 2022).
While the PERK arm of the UPR is pathogenic inMpzS63delmice, IRE1 and ATF6 are implicated in the pathogenesis ofMpzR98Cmice. The precise mechanism is still unclear but the involvement of c-Jun and Krox20 has been proposed. Indeed, c-Jun levels are increased inMpzR98Cmice together with a decrease in Krox20 levels, possibly providing a similar protective role as Sox2 and Id2 upregulation inMpzS63delmice but triggered by the IRE1 and ATF6 arms of the UPR.MpzR98C/+andMpzR98C/R98CSCs show delayed myelination or are arrested at the promyelinating stage, respectively. The authors speculated that signal transduction by IRE1 could activate c-Jun N-terminal kinase, leading to c-Jun phosphorylation and transcription (Patzkó et al., 2012; Saporta et al., 2012) (Figure 1).
Emerging Treatments
Targeting ER stress and the UPR
The accumulation of misfolded proteins in the ER has been considered a therapeutic target to treat CMT1A, CMT1B, and CMT1X. Curcumin is the first compound that has been used to reduce ER stress. Curcumin is an antioxidant and a sarcoplasmic/ER calcium pump inhibitor known to alleviate the retention of misfolded proteins in the ER and therefore to reduce UPR activation (Okamoto et al., 2013). Curcumin treatment has been reported to promote SC differentiation and reduction of UPR activation in a rodent model of CMT1B. For instance,MpzR98Cmice treated with curcumin derivatives performed better at the Rotarod test, had an increased number of largecaliber axons and a better innervation of neuromuscular junctions (Patzkó et al., 2012). Later, a new curcumin formulation has been developed in the form of curcumin-cyclodextrin nanocrystals to increase curcumin’s bioavailability. Treatment of CMT1A rats with this new formulation successfully improved the phenotype, myelination, and nerve conduction velocity (Caillaud et al., 2020). Several studies conducted onMpzS63delmice pointed to the potential of manipulating the PERK arm of the UPR, more specifically stabilizing p-eIF2A, to treat neuropathies in which the UPR is activated (D’Antonio et al, 2013; Sidoli et al., 2016; Scapin et al., 2020). Therefore, attenuating protein translation by modulating the PERK arm could favor the folding and delivery of wild-type P0 to the plasma membrane at the expense of mutant P0 (Bai et al., 2018). Notably, treatment ofMpzS63delmice with salubrinol, a molecule impeding eIF2A dephosphorylation by GADD34 or Sephin 1, a GADD34 holophosphatase inhibitor, reduces P0 retention in the ER and ameliorates disease phenotype (D’Antonio et al, 2013; Das et al, 2015). Moreover, Sephin 1 showed promising results for the treatment of multiple sclerosis (Chen et al, 2019) and its safety and tolerability have been already tested in phase I clinical trials (NCT03610334).
Gene therapy
The very promising results obtained with gene therapy in other neuromuscular diseases such as spinal muscular atrophy (Lowes et al., 2019), have encouraged gene therapy development for CMT as well. One approach consists of the use of adeno-associated viral vectors (AAV). One advantage of using AAVs is that certain serotypes such as AAV9 or AAVRh10 can cross the blood-brain barrier and the blood-nerve-barrier (Liu et al, 2021). In addition, AAVs do not integrate into the genome and provide long-lasting effects (Ghosh et al., 2020). Depending on the type of mutation, AAVs can express either a functional protein or deliver short-hairpin RNA to silence gene expression. Using intraneural delivery of AAV9 coding for a short-hairpin RNA targeted againstPmp22mRNA in CMT1A rats, Gautier et al. (2021) could restore PMP22 levels close to wild-type conditions, improve myelination and ameliorate sensory-motor deficits. In addition, the intraneural delivery route limited off-target effects and greatly improved transduction efficiency. Using the same viral vector, Kagiava et al. (2021) delivered theGjb1gene under the control of theMpzpromoter (active specifically in SCs) by lumbar intrathecal injection inGjb1-null mice. The authors observed a widespread distribution of the vector in myelinating SCs as well as an improvement of the disease phenotype. InGjb1-null mice, intramuscular AAV9-mediated delivery of theNt-3gene, coding for neurotrophin-3, an autocrine growth factor required for SC myelination, also gave promising results (Ozes et al., 2022). A clinical trial aimed at measuring the serum levels of neurotrophin-3 in patients suffering from CMT1A is currently ongoing (NCT05011006).
Two other approaches have been tested to downregulatePmp22mRNA levels. The first one used subcutaneous delivery of antisense oligonucleotides (ASO) in C22 mice and CMT1A rats (Zhao et al., 2018). The treatment successfully delivered ASO to myelinating SCs, normalized PMP22 levels, improved the neuropathy phenotype, and decreased disease progression (Zhao et al., 2018). Notably, transcription profile analysis revealed an increase in the transcription of myelin-related genes together with the downregulation of negative regulators of myelination after ASO treatment (Zhao et al., 2018). Therefore, ASO treatment helped restore SC function. The second very promising approach used the delivery of small interfering RNA conjugated to squalene nanoparticles, which also rescued disease phenotype and increased the levels of Krox20 and SRY-box transcription factor 10 (Sox10) (Boutary et al., 2021).
However, care should be taken when attempting to downregulate PMP22 expression. Indeed, PMP22 levels vary a lot in the disease course, and no clear correlation between PMP22 levels and disease severity has been established in humans. Therefore, more work needs to be carried out to better understand the physiopathology of increased PMP22 expression. In addition, treatment strategies should rather stabilize PMP22 expression over time instead of constantly downregulating PMP22 levels, as this may lead to a similar physiopathology as hereditary neuropathy with pressure palsy (Nobbio et al., 2014; Pantera et al., 2020; Fridman and Saporta, 2021). The challenge for ASO and small interfering RNA injections will be therefore to find the correct dosage and optimal injection intervals (Zhao et al., 2018).
AAV-mediated delivery of wild-type Cx32 may not be sufficient when the mutation has a dominant negative effect. Indeed, upon intrathecal delivery of wild-type Cx32 inGjb1-null mice expressing different CMT1X mutations, wild-type Cx32 was not present in the compact myelin of severalGjb1-null mice expressing Golgi-retained mutants (Kagiava et al., 2018). In this case, the use of clustered regularly interspaced short palindromic repeats/CRISPRassociated protein 9 (CRISPR/CAS9) gene editing technology would be of particular interest since it allows targeted deletions and gene replacement when a homologous repair donor is provided (Liu et al., 2022). CRISPR/CAS9 technology has been used to delete the TATA-box of thePMP22P1 promoter in C22 mice by intraneural delivery. This resulted in effective downregulation of PMP22 expression and preservation of myelin and axon integrity (Lee et al., 2020).
Targeting histone deacetylase activity
Histone deacetylase 2
While current therapeutic approaches aim at downregulating PMP22 expression, Duman et al. (2022) induced remyelination by treating C22 mice with theophylline, an histone deacetylase 2 (HDAC2) activator when used at a low dose (Barnes, 2013). Indeed, recent work by Duman et al. (2020) highlighted the important function of HDAC2 for PNS and central nervous system remyelination. When acetylated, eukaryotic elongation factor 1 A1 (eEF1A1), a major translation elongation factor, translocates to the nucleus, interacts with Sox10 to drag it out of the nucleus and target it to the proteasome for degradation. In contrast, enhancing HDAC2 activity and expression reduces acetylation of eEF1A1, which leads to the return of eEF1A1 to the cytoplasm, prevents Sox10 degradation, and maintains Sox10 on its target genes, thereby increasing remyelination (Duman et al., 2020). Short-term, low-dose theophylline treatment in C22 mice improved the myelination of large-caliber axons and motor functions (Duman et al., 2022). These encouraging results bring a new approach to treating CMT1A disease and its use may be facilitated since theophylline is already an approved drug for asthma and chronic obstructive pulmonary disease at a higher dosage.
Histone deacetylase 3
Recently, Rosenberg et al. (2018) showed that in SCs, histone deacetylase 3 (HDAC3) allows the switch from the adult biogenic state to the homeostatic state, during which the myelin protein synthesis rate decreases to levels needed to maintain the integrity of the myelin sheath during adulthood. In the absence of HDAC3, adult SCs continue to produce myelin at a high rate compared to the differentiation stage (Rosenberg et al., 2018). This study suggests that reducing HDAC3 activity may be beneficial to improve myelination in CMT patients. Indeed, treatment of C3-PMP22 mice with the HDAC3 inhibitor RGFP966 improved axon myelination and electrophysiological parameters and led to the activation of PI3K/AKT and MEK/ERK signaling pathways and to increased myelin protein expression (Prior et al., 2022). Interestingly, Prior et al. (2022) also found more endoneurial macrophages in the nerves of treated animals, suggesting that HDAC3 may also play a role in the modulation of the inflammation process. However, the degree and duration of HDAC3 inhibition have to be carefully monitored. Indeed, mice lacking HDAC3 in SCs present features characteristics of CMT, including motor deficits, limb weakness, myelin outfoldings, and hypermyelination (He et al., 2018; Rosenberg et al., 2018).
Histone deacetylase 6
Upon cellular stress, the heat-shock pathway is activated and assists protein folding or enhances the degradation of misfolded proteins. Chittoor-Vinod et al. (2019) showed that the heat-shock pathway can be activated by inhibiting heat shock protein 90 (HSP90), a molecular chaperone. Furthermore, treatment with the HSP90 inhibitor NVP-AUY922 showed beneficial effectsin vitroandin vivoin C22 mice on myelin synthesis and maintenance and prevented the decline of motor performances (Chittoor-Vinod et al., 2019). Interestingly, HSP90 inhibition or acetylation is known to activate the expression of heat shock protein 70 and other chaperones and to reduce PMP22 aggregates (Kovacs et al., 2005; Chittoor-Vinod et al., 2015). C22 mice treated with CKD-504, an histone deacetylase 6 (HDAC6) inhibitor, show increased levels of acetylated HSP90 together with heat shock protein 70 upregulation. The authors suggested that the increase of myelinated axons as well as the improved behavioral and electrophysiological performances of C22 mice treated with CKD-504 is due to the activation of chaperonemediated PMP22 folding (Ha et al., 2020). Of note, HDAC6 inhibitors were also previously shown to improve axonal transport and function in axonal CMT2 models (reviewed in Rossaert and Van Den Bosch, 2020).
Neuregulin 1-type III and -type I
NRG1-type III activity is regulated by two secretases: the beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) and tumor necrosis factoralpha-converting enzyme (TACE). By different extracellular cleavages, BACE1 enhances and TACE inhibits NRG1-type III activity (Pellegatta and Taveggia, 2019). Recently, Scapin et al. (2019) overexpressed NRG1-type III inMpzS63delmice and observed an amelioration of neurophysiological parameters and increased myelin thickness. Surprisingly, Krox20 and myelin protein levels remained unaffected, but myelin lipid content was increased. This indicates that NRG1-type III overexpression acts in a Krox20-independent manner and determines myelin sheath thickness by increasing lipid content (Scapin et al., 2019). This contrasts with the activation of Krox20 and subsequently of myelin protein and lipid genes upon NRG1 signaling during SC development (Kao et al., 2009). The modulation of NRG1-type III signaling via pharmacological inhibition of TACE has also been used inMpzS63delmice using BMS-561392, which has already been used in phase II clinical trials for the treatment of rheumatoid arthritis (Moss et al., 2008; Scapin et al., 2019). Unfortunately, the drug was unable to cross the blood-brain and blood-nerve barrier, and a new formulation or administration route would be necessary for further investigations in CMT1B models or patients (Scapin et al., 2019). However, treatment ofMpzS63deldorsal root ganglia explants with BMS-561392 improved myelination.
Besides NRG1-type III, SC-derived NRG1-type I is also crucial for nerve regeneration and remyelination after nerve injury (Stassart et al., 2013). In line with this observation, treatment with recombinant human NRG1 in CMT1A rats early after birth restores the balance between PI3K/AKT and MEK/ERK signaling pathways, favors SC differentiation, myelination, and downregulates SC immature markers. Furthermore, CMT1A rats show better motor performances and an increased number of myelinated axons. In line with the supportive functions of NRG1-type I in CMT1A rats during early postnatal stages, treatment with recombinant human NRG1 could mimic axonal NRG1-type III function early after birth and promoted myelination (Fledrich et al., 2014). These results and the fact that recombinant human NRG1 has already been tested in phase II clinical trials to treat cardiovascular diseases (reviewed in Geissler et al., 2020) encourage further investigations into the use of recombinant human NRG1 in patients suffering from CMT1A. However, the improvements are only visible when the animals are treated early after birth. Therefore, this therapeutic approach should be applied during early childhood before disease onset. Indeed, NRG1-type I is upregulated in adult CMT1A rats and C61 mice, and genetic ablation of NRG1-type I in SCs ameliorates disease phenotype with suppression of hypermyelination and of onion bulb formation. This indicates that the supportive functions of NRG1-type I in SCs are restricted to early postnatal development and the treatment of adult CMT1A patients would require another therapeutic approach, such as potentially pharmacological inhibitors of ErbB2 receptors (Fledrich et al., 2014, 2019).
Other therapies
Currently, a very promising treatment for CMT1A is PTX3003, a cocktail composed of baclofen, naltrexone, and D-sorbitol. Administration of PTX3003 in CMT1A rats induces SC differentiation and myelination, reducesPMP22mRNAs levels, restores the balance between PI3K/AKT and MEK/ERK signaling pathways, and improves the phenotype (Chumakov et al., 2014; Prukop et al., 2019). However, myelin thickness and the number of myelinated axons did not improve. The beneficial effects of PTX3003 treatment are rather due to the increase of functional neuromuscular junctions and muscle innervation observed in CMT1A rats, independently of myelination (Prukop et al., 2020). Phase II and III clinical trials (NCT02579759) have demonstrated the safety and tolerability of PTX3003. In addition, CMT Neuropathy Score, Overall Neuropathy Limitations Scale and 10-minute walk test were significantly improved in CMT1A patients receiving the higher dose of PTX3003 (Attarian et al., 2014; Vita et al., 2019).
Another potential pathomechanism in CMT1A may be the increased expression of the P2X purinoreceptor 7 (P2X7) and the subsequent increase of intracellular Ca2+concentration in SCs of CMT1A rats (Nobbio et al., 2009). Sociali et al. (2016) tested the therapeutic potential of the P2X7 antagonist A438079in vivoin CMT1A rats. Upon A438079 treatment, hind limb muscle strength, electrophysiological and morphological features as well as SC differentiation were improved in CMT1A rats (Sociali et al., 2016). In addition, A438079 was already approved in a phase II clinical trial for the treatment of rheumatoid arthritis (Keystone et al., 2012). The CSF-1 receptor has also been considered a therapeutic target. Indeed, treatment of CMT1X and CMT1B mouse models with the CSF-1 receptor inhibitor PLX5622 reduced macrophage number, improved axonal integrity, and ameliorated the phenotype (Klein et al., 2015). Finally, transcriptome analysis of CMT1A rats revealed a transcriptional dysregulation of genes coding for myelin lipid biosynthesis (Fledrich et al., 2018). Since myelin is composed of 70% lipids and PMP22 plays an important role in lipid biosynthesis, trafficking, and localization (see CMT1A section), modulating lipid biosynthesis appears to be an interesting therapeutic option. By supplementing the diet of CMT1A rats with phosphatidylcholine and phosphatidylethanolamine, Fledrich et al. (2018) observed an increase in the number of myelinated axons and an amelioration of nerve function.
The recent therapeutic strategies for the demyelinating and dysmyelinating forms of CMT are summarized in Table 1.
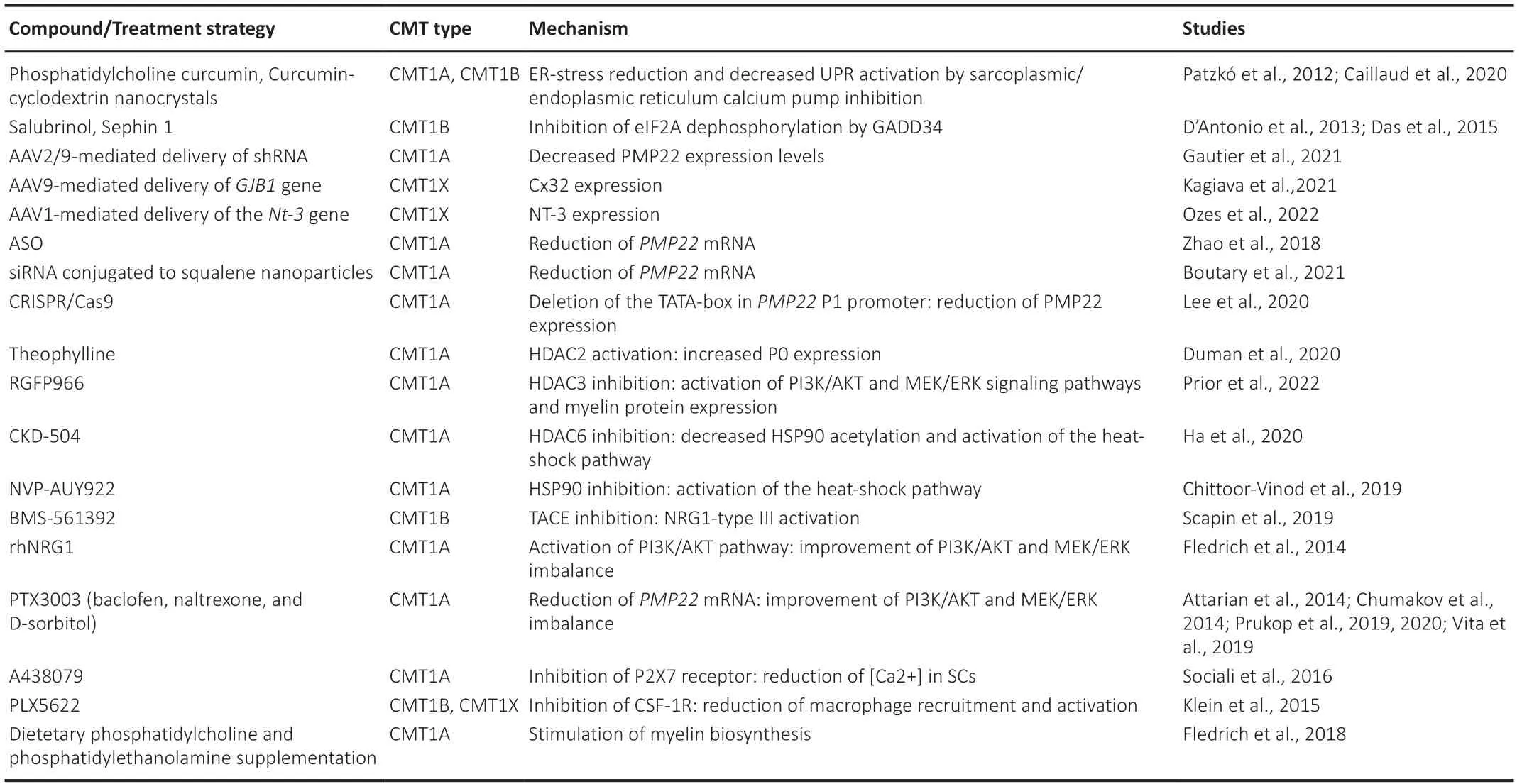
Table 1 |Summary of therapeutic strategies for demyelinating and dysmyelinating Charcot-Marie-Tooth disease
Conclusion
The recent advances in CMT research allowed expanding the knowledge of the molecular mechanisms underlying the demyelinating or dysmyelinating forms of CMT, which appear varied and complex. These mechanisms share similarities with SC response to injury and the discoveries on SC conversion into repair SCs after lesion may be of great help to better understand SC biology during CMT (this review and Arthur-Farraj and Coleman, 2021), as exemplified by the expression of NRG1-type I and its function in inducing hypermyelination and onion bulb formation in CMT1A (Stassart et al., 2013; Fledrich et al., 2019), the activation of MEK/ERK signaling pathway (Fischer et al. 2008; Groh et al, 2010, 2015; Kohl et al., 2010; Fledrich et al., 2014, 2019; Scapin et al., 2020), the expression of SC immature and repair markers in CMT1A, CMT1B and CMT1X (Patzkó et al., 2012; Saporta et al., 2012; D’Antonio et al., 2013; Fledrich et al., 2014; Groh et al., 2015; Florio et al. 2018; Scapin et al., 2020) and the inflammatory process in CMT1X (Fischer et al., 2008; Groh et al., 2010, 2015; Kohl et al., 2010; Martini et al., 2013). Our knowledge on SC response after injury could therefore allow finding interesting strategies to treat CMT. In addition, more work aimed at better understanding the origin and physiopathology of onion bulb formation, contributing to hypertrophic neuropathy and a hallmark of CMT1A, may improve our knowledge on cellular and molecular mechanisms underlying disease phenotype (Fledrich et al., 2019; Tracy et al., 2019). The discoveries regarding the implication of the ERstress and UPR in CMT1 pathogenesis have also shed light on new therapeutic targets. However, the genetic diversity of CMT may not allow finding a unifying treatment for all CMT forms. Furthermore, regarding all the different gene variants, the treatment would rather be allele-specific. Therefore, it is crucial to decipher the pathomechanisms of the different alleles and to adapt the rodent models accordingly. Notwithstanding these challenges, the recent therapeutic approaches discussed above showed promising results in preclinical studies and some of them are already being tested in phase I, II, or III clinical trials with very encouraging results. Furthermore, advances in gene replacement therapy offer a new perspective for allele-specific curative treatments. However, to detect a potential benefit of these treatments, it is of utmost importance to properly design clinical trials and develop appropriate biomarkers, in particular in the case of slow disease progression such as in CMT1A patients (Svaren et al., 2019; Rossor et al., 2020).
Author contributions:CJ and NH designed the manuscript. NH searched and analyzed the litterature, wrote the manuscript, and designed the figure and the table. CJ corrected and revised the manuscript. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Data availability statement:No additional data are available.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Mesenchymal stem cells, extracellular vesicles, and transcranial magnetic stimulation for ferroptosis after spinal cord injury
- Inducing prion protein shedding as a neuroprotective and regenerative approach in pathological conditions of the brain: from theory to facts
- Use of mesenchymal stem cell therapy in COVID-19 related strokes
- Brain organoids are new tool for drug screening of neurological diseases
- Emerging roles of astrocytes in blood-brain barrier disruption upon amyloid-beta insults in Alzheimer’s disease
- External anal sphincter electromyography in multiple system atrophy: implications for diagnosis, clinical correlations, and novel insights into prognosis