
Reviving the use of inhibitors of matrix metalloproteases in spinal cord injury: a case for specificity
2023-02-13 12:41ZubairAhmed
中国神经再生研究(英文版) 2023年9期
Zubair Ahmed
Abstract At present, there are no restorative therapies in the clinic for spinal cord injury, with current treatments offering only palliative treatment options. The role of matrix metalloproteases is well established in spinal cord injury, however, translation into the clinical space was plagued by early designs of matrix metalloprotease inhibitors that lacked specificity and fears of musculoskeletal syndrome prevented their further development. Newer, much more specific matrix metalloprotease inhibitors have revived the possibility of using these inhibitors in the clinic since they are much more specific to their target matrix metalloproteases. Here, the evidence for use of matrix metalloproteases after spinal cord injury is reviewed and researchers are urged to overcome their old fears regarding matrix metalloprotease inhibition and possible side effects for the field to progress. Recently published work by us shows that inhibition of specific matrix metalloproteases after spinal cord injury holds promise since four key consequences of spinal cord injury could be alleviated by specific, next-generation matrix metalloprotease inhibitors. For example, specific inhibition of matrix metalloprotease-9 and matrix metalloprotease-12 within 24 hours after injury and for 3 days, alleviates spinal cord injury-induced edema, blood-spinal cord barrier breakdown, neuropathic pain and restores sensory and locomotor function. Attempts are now underway to translate this therapy into the clinic.
Key Words: axon regeneration; blood-spinal cord barrier; edema; functional recovery; matrix metalloprotease-9; matrix metalloproteses-12; pain; spinal cord injury
Introduction
Spinal cord injury (SCI) is a devastating condition, often causing permanent loss of function and significant disability (Wilson et al., 2012). At present, there are no fully restorative treatments available for SCI and hence this remains an area of urgent medical need. Treatments such as Lyrica and Medrol, only offer symptomatic relief to patients from neuropathic pain but do not address the underlying mechanisms that cause neuropathic pain or inflammation (Tong et al., 2021). A fully restorative therapy for SCI would therefore promote the protection of neurons and glia from further damage, neutralize inhibitory molecules, inhibit the glial scar, replace lost neurons, and promote axon regeneration and appropriate synapse formation.
Matrix metalloproteases (MMPs) are zinc-dependent enzymes that are involved in a variety of proteolytic events during development, wound healing, and repair processes (Sternlicht and Werb, 2001). For several decades, the role of MMPs has been investigated in the pathogenesis of neurodegenerative diseases and models of central nervous system (CNS) injury, including SCI and traumatic brain injury (Yong, 2005; Zhang et al., 2010). In SCI, MMPs contribute to the degradation of the blood-spinal cord barrier (BSCB), oxidative stress, demyelination, and a progressive neuroinflammatory response (Noble et al., 2002; Wells et al., 2003; Zhang et al., 2011).
The MMPs are a family of 23 human or 24 murine members and can be subdivided into eight different types based on their domain structure and substrate specificity (Yong, 2005; Page-McCaw et al., 2007; Zhang et al., 2011; Table 1). MMPs are initially produced as inactive zymogens and require catalytic activation where the propeptide domain is cleaved and thus exposing the active catalytic site. MMPs are regulated at both the transcriptional and post-translation levels and their activity is regulated by four endogenous inhibitors of metalloproteases, called tissue inhibitors of MMPs (TIMPs) (Sternlicht and Werb, 2001). TIMPs can sequester MMP activity by binding to their catalytic sites and thus precisely control their activity and limit extracellular matrix (ECM) degradation (Sternlicht and Werb, 2001; Crocker et al., 2004).
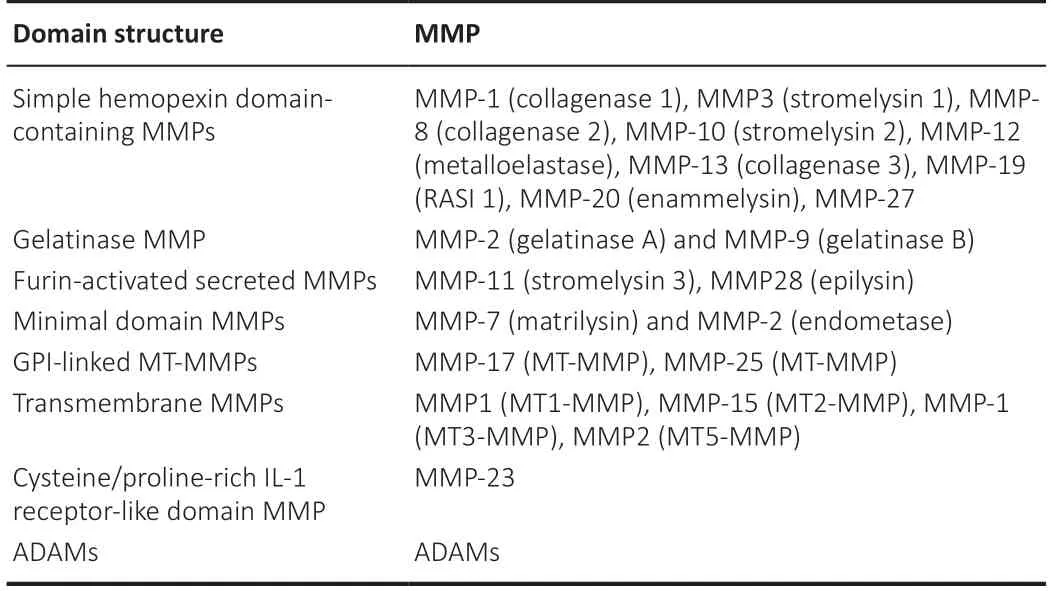
Table 1 |Classification of MMPs based on domain structures
Search Strategy and Selection Criteria
The search strategy and selection criteria were limited to articles published in peer-reviewed journals articles. A literature review of articles was conducted by searching PubMed and Web of Science databases, updated until August 2022, searching for the following topics: matrix metalloproteases and/or MMP and spinal cord injury, matrix metalloproteases and/or MMP and SCI, matrix metalloprotease and/or MMP-9 and spinal cord injury, matrix metalloprotease and/or MMP-12 and spinal cord injury, matrix metalloprotease and/or MMP inhibitors and spinal cord injury, matrix metalloprotease and/or MMP-9 inhibitor and spinal cord injury, matrix metalloprotease and/or MMP-12 inhibitor and spinal cord injury. The selected articles focused on MMP expression and MMP inhibition in spinal cord injury. Human and animal studies were all screened.
Matrix Metalloproteases in Spinal Cord Injury
A number of studies have profiled the expression of MMPs after SCI. The first MMPs to be studied in SCI were gelatinases MMP-9 and MMP-2. It was reported that MMP-9 activity was observed within 12–24 hours postinjury whilst MMP-2 rose significantly by 5 days after contusion injury in the rat (de Castro et al., 2000). These results were later confirmed in other species and models of SCI, including hemisection and compression in a flurry of studies. For example, zymography and western blot assay showed transient increases in MMP-9 at 24 hours post injury with a gradual increase in MMP-2 (Duchossoy et al., 2001; Xu et al., 2001; Goussev et al., 2003; Hsu et al., 2006; Yu et al., 2008). In a compression model of SCI, mRNAs for MMP-9, MMP-3, MMP-7, MMP-10, MMP-11, MMP13, MMP-19, and MMP-20 were significantly upregulated within 24 hours whereas MMP-2, MMP-12, and MMP-13 upregulation was delayed until 5 days after SCI (Wells et al., 2003; Veeravalli et al., 2009; Table 2).
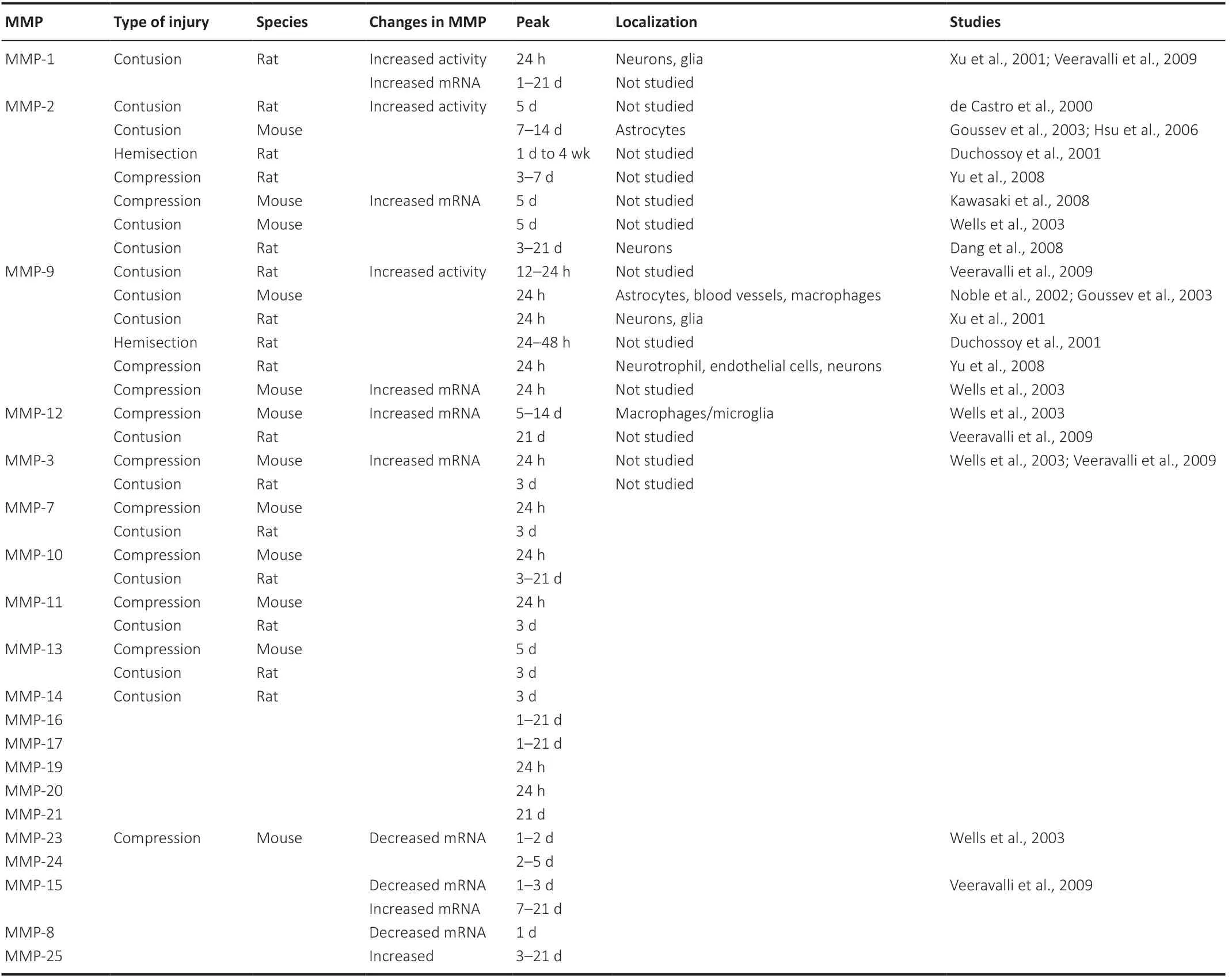
Table 2|Summary of MMPs in experimental spinal cord injury models
After SCI, MMP-1 and MMP-9 were both localized in glia and neurons (Xu et al., 2001), whilst MMP-9 was also observed in blood vessels, neutrophils, and macrophages (Xu et al., 2001; Noble et al., 2002; Yu et al., 2008). MMP-2 not only localizes to neurons and astrocytes but also to microglia and macrophages (Wells et al., 2003; Hsu et al., 2006; Veeravalli et al., 2009). Other MMPs were also localized in the spinal cord after injury but their temporal and cellular localization varied after SCI and related to the type and severity of the injury.
Matrix Metalloproteases and Their Inhibitors in Spinal Cord Injury
In the acute phase after CNS injury, MMPs largely play a number of detrimental roles such as degradation of the basal lamina, disruption of BSCB, oxidative stress, demyelination, leukocyte infiltration, and progressive neuroinflammation (Noble et al., 2002; Wells et al., 2003; Hsu et al., 2006; Zhang et al., 2011). In the chronic phase, however, MMPs play a beneficiary role in modulating the inhibitory glial scar, promoting cell survival, supporting axonal regeneration, and attenuating mechanical allodynia (Chang and Werb, 2001; Duchossoy et al., 2001; Yong, 2005; Zhao et al., 2006). Therefore, inhibiting MMP activity is a complex process and requires intricate knowledge of the timeline of MMP activities after SCI.
Inflammation and Blood-Spinal Cord Barrier Breakdown
MMPs facilitate the extravasation of inflammatory cells into the injured spinal cord and contribute to the breakdown of the BSCB. When infiltrating leukocytes migrate across the vascular wall, they secrete MMPs which then degrade tight junction proteins since zonulae occludens-1, VE-cadherin, and occludins are substrates for MMP-2, -3, -7, and -9 (Asahi et al., 2001; Caron et al., 2005; Yang et al., 2007; Buhler et al., 2009). The target of MMPs also included proteins found in the basal lamina such as laminin, fibronectin, and heparan sulfate (Rosenberg and Yang, 2007).MMP-9 mediates acute SCI pathogenesis in the injured spinal cord, derived primarily from infiltrating leukocytes since depleting leukocytes reduces MMP-9 activity in injured tissues (de Castro et al., 2000). MMP-9 expression (in both rodents and humans) peaks within one day after SCI and is detected in glia, macrophages, neutrophils, and vascular element in the SCI 24 hours after injury (Noble et al., 2002; Goussev et al., 2003; Buss et al., 2007). Excessive activity of MMP-9 in the acute phase of SCI disrupts the BSCB, edema, excitotoxicity, infiltration of leukocytes, mitochondrial dysfunction, apoptosis, demyelination of neurons, increase in inflammatory responses and astrogliosis (Noble et al., 2002). Disruption of BSCB after SCI is maximal 24 hours after injury, coinciding with the peak activity of MMP-9 in the injured spinal cord (Noble et al., 2002). Indeed, BSCB disruption was reduced in MMP-9 KO mice and mice treated with the broad-spectrum MMP inhibitor, GM6001 (Noble et al., 2002). Moreover, treatment with SB-3CT, a gelatinase inhibitor, at 2 hours before SCI, reduced MMP-9 activity and BSCB disruption, ultimately decreasing apoptosis (Yu et al., 2008).
Similar findings were also observed in MMP-12 KO mice, where greater stabilization of the BSCB was observed. Indeed, MMP-12 (macrophage metalloelastase) is required for the migration of blood-borne macrophages across endothelial cell basement membranes and into areas of inflammation (Shipley et al., 1996; Wells et al., 2003). MMP-12 is also expressed by macrophages and increases 182-fold in a compression SCI model, perpetuating the inflammatory response and acute edema and promoting the development of the secondary injury response (Wells et al., 2003, 2005).
Oxidative Stress and Apoptosis
MMPs are regulated by oxidative stress and reactive oxygen species such as nitric oxide and hypochlorous acid increase MMP-9 and MMP-9 activity and decrease TIMPs (Rajagopalan et al., 1996; Morita-Fujimura et al., 2000). Increased levels of oxidative stress after SCI lead to MMP-9 upregulation and BSCB breakdown as well as apoptosis via neurotoxicity (Yu et al., 2008). In support of this, transgenic rats that overexpress the antioxidant enzyme superoxide dismutase 1, showed that oxidative stress and MMP-9-mediated BSCB breakdown and apoptosis were all attenuated (Morita-Fujimura et al., 2000; Sugawara et al., 2002). MMP-2 contributes to apoptosis and is upregulated after injury, coinciding with glial and neuronal apoptosis. MMP-2 therefore has been suggested to mediate apoptosis after SCI since the suppression of MMP-2/MMP-9 reduced the levels of apoptosis in glia and neurons, which may promote long-term recovery (Dang et al., 2008).
Importantly, these studies demonstrate that MMPs direct disruption of the BSCB, infiltration of leukocytes and apoptosis after SCI. Since early and short-term (for up to 5 days after SCI) inhibition of MMPs stabilized the BSCB, reduced infiltration of leukocytes may confer acute and longterm neuroprotection, suggesting that acute inhibition of MMPs may be a promising strategy for SCI.
Matrix Metalloproteases in Glial Scar Formation
A glial scar at the lesion site normally forms after SCI, containing axon growth inhibitory molecules, ECM molecules, microglia/macrophages, and reactive astrocytes and ECM molecules such as chondroitin sulphate proteoglycans (CSPG). Increased expression of CSPGs results from the interaction of astrocytes, oligodendrocytes, and macrophages, which in turn inhibits axon regeneration (Bradbury et al., 2002; Jones et al., 2002). MMPs degrade the core protein of a variety of inhibitory molecules including CSPGs, Nogo, and tenascin-C, whilst CSPGs such as tenascin-C, brevican, neurocan, NG2, phosphacan, and versican are degraded by MMP activity (Pizzi and Crowe, 2007). Therefore, MMPs support axonal regeneration in the injured CNS by degrading CSPGs and other inhibitory molecules (Yong, 2005; Pizzi and Crowe, 2007).
MMPs also facilitate the migration of astrocytes since gelatinase activity is correlated with scar formation (Duchossoy et al., 2001).In vitroassays demonstrate that astrocytes derived from MMP-9 KO mice or astrocytes treated with an MMP-9 inhibitor both attenuate their migration (Hsu et al., 2008). Consistent with this observation, the glial scar is abrogated after SCI in MMP-9 KO mice along with reduced CSPG immunohistochemistry (Hsu et al., 2008). On the other hand, MMP-2 KO mice showed increased immunoreactivity for CSPG, fewer serotonergic fibers and significantly reduced motor recovery compared to wild-type mice, suggesting reduced axonal sprouting across the lesion site and the important role that MMP-2 plays in promoting functional recovery after SCI (Hsu et al., 2006). MMPs also play a role in axonal dieback since inhibition of MMP-9 prevented macrophage-mediated axon retraction (Busch et al., 2009). These studies suggest that MMPs are critical to the formation of a glial scar but can degrade inhibitory molecules and cleave ECM proteins that sequester growth factors and hence support function recovery (Yong et al., 2001; Pizzi and Crowe, 2007).
Matrix Metalloproteases in Neuropathic Pain
Neuropathic pain is common in as many as 85% of SCI patients in dermatomes above and below the lesion, manifesting in allodynia and hyperalgesia (Siddall et al., 2003) but the mechanisms causing neuropathic pain remains unanswered. Neuropathic pain is normally studied using the peripheral nerve injury model where neuropathic pain can be modeled in the absence of damage to central pathways. However, studies suggest that the mechanisms causing neuropathic pain after SCI and peripheral nerve injury are similar (Detloff et al., 2008). In peripheral nerve injury, MMPs both induce and maintain neuropathic pain, primarily regulated by MMP-9 and MMP-2. MMP-9 levels rise rapidly within 24 hours after injury whilst MMP-2 levels are delayed until day 7 but persist for up to 21 days (Kawasaki et al., 2008). The differential time course of MMP-9 and MMP-2 in the development and maintenance of neuropathic pain after injury suggests a role for MMP-9 and MMP-2 but a direct role is yet to be established.
It is thought that mechanical injury to axons causes Schwann cells to secrete MMP-9 which then activates the infiltration of macrophages and stimulates the degradation of myelin proteins such as myelin basic protein (Chandler et al., 1995; Kobayashi et al., 2008). This causes exposure of the bare nerve endings, resulting in increased sodium channel expression and ectopic hyperexcitability of afferents (Devor, 2006). Action potentials now outlast the stimulus and create central sensitization, a common mechanism that is found in the transmission of neuropathic pain.
It has long been established that proinflammatory cytokines and growth factors also regulate neuropathic pain since they mediate the expression of gelatinases in the CNS. For example, tumor necrosis factor-α or interleukin-1β (IL-1β) induces MMP-9 expression by Schwann cells, leading to neuronal sensitization (Chattopadhyay et al., 2007; Schafers and Sorkin, 2008). Deletion of MMP-9 reduces pain behaviors after peripheral nerve injury, whilst blocking IL-1β signaling with a neutralizing antibody-prevented allodynia (Chattopadhyay et al., 2007; Kawasaki et al., 2008). Conversely, intrathecal injection of MMP-9 increased the levels of allodynia with increased cleavage of IL-1β and microglial activation in the dorsal horn and colocalized with activated p38 MAP kinase (Kawasaki et al., 2008). MMP-2 also cleaves IL-1β and activates spinal astrocytes at later time points, potentially maintaining neuropathic pain (Kawasaki et al., 2008). This suggests that MMP-9 and MMP-2 are intricately related to IL-1β, which may be a potential downstream regulator of neuropathic pain (Figure 1).Apart from gene deletion (Table 3), short interfering RNA (siRNA) and peptide inhibitors to MMP-9 and MMP-2 have been used and demonstrated to be effective in reducing allodynia in the peripheral nerve injury model. Daily injections of broad-spectrum gelatinase inhibitor, GM6001, resulted in immediate and sustained attenuation of allodynia, preserving myelin basic protein, reducing infiltration of macrophages and reducing glial activation in the dorsal horn (Kobayashi et al., 2008). Likewise, treatment with siRNA to MMP-9 prevented the onset of allodynia whilst MMP-2 siRNA also attenuated allodynia (Kawasaki et al., 2008). Administration of TIMP-1 and TIMP-2 led to a greater reversal of allodynia but the effects were transient and lasted 3 to 24 hours (Kawasaki et al., 2008). Inhibition of MMP-9 using Inhibitor-I delayed the onset of allodynia by 6 days whilst inhibition of MMP-2 using Inhibitor III attenuated allodynia for 10 days (Kawasaki et al., 2008) suggesting that MMP-9 and MMP-2 inhibition can attenuate neuropathic pain.

Table 3 |KO animals and their effects in SCI
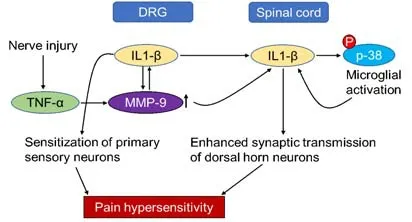
Figure 1|Schematic to show mechanisms of MMP-induced neuropathic pain.
Matrix Metalloproteases in Human Spinal Cord Injury
Although the majority of the data on MMPs in SCI is from animal studies, there are a small number of studies in humans that corroborate some of the findings. For example, in post-mortem spinal cord MMP-9 was found at 2 days after injury (which was the earliest time studied) at the lesion site and localized in neutrophils and macrophages (Buss et al., 2007) whilst another study localized MMP-9 in neutrophils with peak expression at 4 hours – 3 days, in agreement with rodent studies (Fleming et al., 2006). However, MMP-12 showed a delayed expression of 24 days, far greater than the 5-day peak expression observed in mouse/rat studies (Buss et al., 2007). This may be due to an artifact arising from the usage of post-mortem spinal cord for experiments or could be a discrepancy arising from mouse and humandisease models. Although an exploratory study suggested using serum levels of MMP-8 and MMP-9 as early markers for remission, the sample sizes were too low to make definitive conclusions (Moghaddam et al., 2017). Further studies into the temporal expression of MMPs after human SCI are required but are obviously difficult to perform. However, serum and cerebrospinal fluid levels of MMPs will give us an indication of what is happening in terms of MMP activation after SCI in humans.
Matrix Metalloprotease Inhibitors
The search for MMP inhibitors began over three decades ago (Table 4) but initial clinical trials were disappointing and painted a negative picture of MMPs as therapeutic targets. With an improved understanding of the biology of MMPs, it emerged that clinical trials were conducted prematurely. Better-designed MMP inhibitors had desirable selectivity’s and improved pharmacokinetic profiles, resulting in lower toxicity. Selective MMP inhibitors demonstrated that MMP-2, MMP-9, MMP-13, and MT1-MMP were not involved in musculoskeletal syndrome that plagued early, broad-spectrum MMP inhibitors. Musculoskeletal syndrome appeared to be caused by offtarget effects of inhibiting not just one MMP but a combination of MMPs and/or possibly other related enzymes (Peterson, 2006).

Table 4 |MMP inhibitors and their use after SCI
The first generation of peptidomimetics MMP inhibitors was broad-spectrum, containing a hydroxamate moiety that chelated catalytic zinc and inactivated the protein such as batimastat, marimastat, and ilomastat. The hydroxamate moiety was readily metabolized and non-specifically inhibited ADAMs as well as producing musculoskeletal syndrome (Peterson, 2004; Jacobsen et al., 2010). Other inhibitors include those that contain a mercaptoacyl as a zincbinding group (Rebimastat), and tanomastat has a zinc-binding carboxylate group. However, as these are zinc chelators they target gelatinases as well as other zinc-dependent enzymes, including other MMPs. Doxycycline and Minocycline are tetracycline-based inhibitors with broad-spectrum MMP inhibitory activity (Romero-Perez et al., 2008), whilst bisphosphonates and carbamoyl phosphobates also inhibit various MMPs (Teronen et al., 1999; Matziari et al., 2004).
SB-3CT was the first mechanism-based inhibitor of gelatinase where the inhibitor-enzyme complex underwent a requisite conformational change that did not readily reverse. SB-3CT is selective for MMP-2 and MMP-9 but does not inhibit other MMPs except for MMP-14 (Toth et al., 2000). SB-3CT showed enhanced efficacy than precious MMP inhibitors but it was rapidly metabolized to a more active gelatinase inhibitor than the parent molecule, limiting its use (Lee et al., 2007). Although these early MMP inhibitors displayed high affinityin vitro, the first generation of MMP inhibitors failed in clinical trials due to their low selectivity and nonspecific inhibition of MMPs and non-related proteins that can be affected by the metal chelating groupsin vivo(Overall and Kleifeld, 2006; Fingleton, 2008; Lopez-Otin et al., 2009).
Better designs of MMP inhibitors soon followed with the targeting of inhibitory sites outside the conserved catalytic cleft, the exosites, which produced selective MMP inhibitionin vivoand thus revived the field’s interest to selectively control the pathophysiological activity of these enzymes (Turk, 2006; Sela-Passwell et al., 2010; Deu et al., 2012). This led to the development of macromolecule inhibitors, which were inhibitory antibodies against various MMPs beyond the enzyme catalytic domain. These antibodies were much more specific and highly selective but still retained the homeostatic activity of other MMP family members. Highly selective MMP-9 inhibitors such as AB0041 and AB0046 were developed either as full-length human MMP-9 or a mouse Pro-Cat construct (Marshall et al., 2015). Other inhibitory antibodies such as SDS3, an MMP-2/-9 inhibitor soon followed (Sela-Passwell et al., 2011) together with the first anti-MMP-9 antibody (REGA-3GL12) (Paemen et al., 1995).There are several clinical trials listed on ClinicalTrial.gov (last accessed 30/05/2022) evaluating MMP inhibitors in a variety of conditions such as cancer, glioblastoma, open-angle glaucoma, asthma, and chronic obstructive pulmonary disease. With the advent of much more specific MMP inhibitors, the time is now right to let go of the dogma of viewing MMP inhibition as intractable.
Inhibition of Matrix Metalloprotease-9 and Matrix Metalloprotease-12 Using AZD1236 to Improve Function after Spinal Cord Injury
AZD1236 is a potent and reversible inhibitor of MMP-9 and MMP-12, with an IC50of 4.5 and 6.1 nM, respectively. It has > 10-fold selectivity to MMP-12 and MMP-13 and > 350-fold selectivity to other members of the MMP family. AZD1236 was originally developed by AstraZeneca for use in chronic obstructive pulmonary disease and has been tested in single doses up to 1500 mg and 500 mg QD for 13 days, whilst COPD patients were dosed for 6 weeks with 75 mg BID (Magnussen et al., 2011; Dahl et al., 2012). We re-purposed AZD1236 in SCI and showed unprecedented benefits (Ahmed et al., 2022).
By critically analyzing the known pathophysiology associated with SCI, it is evident that excessive MMP activity, in particular, MMP-9 and MMP-12, contribute to four of the key pathological drivers of SCI, namely; disruption of the BSCB, edema, neuropathic pain and loss of motor/sensory function. We found that MMP-9 levels and their activity in the spinal cord, serum, and cerebrospinal fluid peaked within the first 24 hours, whilst MMP-12 peaked 5 days post-SCI (Ahmed et al., 2022). Inhibition of MMP-9 and MMP-12 activity using oral or intrathecal delivery of AZD1236, attenuated the SCI-induced rise in water content in the spinal cord by 98%; an effect that was directly related to inhibition of both MMP-9 and MMP-12. Measures of proinflammatory pain markers, BSCB breakdown, and scarring at the lesion site were also suppressed by 80%, 75%, and 80%, respectively. Inhibition of MMP-9 and MMP-12 promoted axon regeneration and protected against SCI-induced sensory and locomotor deficits such that animals behaved as uninjured sham controls by 3 weeks post-injury. Suppressed microglial/macrophage activation and reduced scar-derived inhibitors of axon growth at the lesion site, including Semaphorin-3A and CS-56, were also reduced further contributing to the promotion of axon regeneration after SCI (Bradbury et al., 2002).
Interestingly, the reduction of SCI-induced edema was reliant on the combined suppression of both MMP-9 and MMP-12, since the suppression of either of these individually had a suboptimal impact on spinal cord edema (Ahmed et al., 2022). Indeed, the clinical outcomes after SCI are greatly affected by spinal cord edema and if left unchecked this can lead to further damage and death, posing a significant challenge to neurosurgeons (Rutges et al., 2017).
Our findings, therefore, demonstrate that selective short-term inhibition of MMP-9 and MMP-12 has the potential to offer previously unparalleled levels of protection against secondary damage to the spinal cord as well as the dramatic potential for recovery. The effects of AZD1236 after SCI show that the beneficial effects of MMP inhibition lie primarily in their selectivity and this plagued early MMP inhibitor designs. However, with advances in protein engineering and new technologies, it is possible to design next-generation MMP inhibitors with unparalleled selectivity and specificity such that old fears regarding MMP inhibition and possible side effects need to be overcome for the field to progress further. Our work in the spinal cord brings hope that MMP inhibition to target specific diseases may become a possible reality for future clinical translation.
Author contributions:ZA conceptualized and conducted the literature review, wrote the manuscript and created the Figures and Tables. ZA edited text and figures, revised the whole manuscript, and approved the final version of the manuscript for publication.
Conflicts of interest:The author is an inventor on a patent related to MMP-9 and MMP-12 inhibitors in neurological diseases.
Data availability statement:No additional data are available.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Mesenchymal stem cells, extracellular vesicles, and transcranial magnetic stimulation for ferroptosis after spinal cord injury
- Inducing prion protein shedding as a neuroprotective and regenerative approach in pathological conditions of the brain: from theory to facts
- Use of mesenchymal stem cell therapy in COVID-19 related strokes
- Brain organoids are new tool for drug screening of neurological diseases
- Emerging roles of astrocytes in blood-brain barrier disruption upon amyloid-beta insults in Alzheimer’s disease
- External anal sphincter electromyography in multiple system atrophy: implications for diagnosis, clinical correlations, and novel insights into prognosis