
超高效液相色谱-串联质谱法直接同时测定牛乳中草铵膦及其代谢物
2023-02-07 06:48:56张立佳刘丽君谢瑞龙吕志勇李翠枝
食品科学 2023年2期
张立佳,刘丽君,汪 洋,文 静,莫 楠,谢瑞龙,吕志勇,李翠枝
(内蒙古伊利实业集团股份有限公司,内蒙古 呼和浩特 010110)
草铵膦是我国允许使用的除草剂。中国作为全球农业大国,草铵膦转基因技术推广和应用,已经成为全球第二大转基因作物除草剂[1]。草铵膦是属于新型广谱长效触杀型化学除草剂,具有化学活性高、低毒、无污染、易降解、对作物安全、环境友好和除草快捷等特点[2]。尽管草铵膦是属于广谱非持久性农药,但如果放任其大量的、无约束的长期使用,则仍是有一定可能在农作物上产生残留,再通过食物链进入人类的餐桌,影响人体健康。草铵膦也可以在动物体内代谢为3-(甲基膦基)丙酸和N-乙酰基草铵膦。大量研究发现吸入低剂量的草铵膦可以伤害新生儿和动物幼崽大脑的发育,高剂量可以导致过敏、高烧、精神紊乱、流涕、抽搐等中毒症状,此外,该药物的残留可以升级为食品安全事件,扰乱国际进出口贸易[3-4]。世界各国已制定了草铵膦最大残留限量的规定,美国和日本限量值为0.05~6.00 mg/kg[5],加拿大限量值为0.10~2.00 mg/kg[6],国际食品法典规定0.02~8.00 mg/kg[7],欧盟规定0.10~5.00 mg/kg[8],GB 2763—2021《食品中农药最大残留限量标准》规定为0.01~0.50 mg/kg,其中牛乳中最大残留限量为0.02 mg/kg[9],并定义以草铵膦及其3-(甲基膦基)丙酸和N-乙酰基草铵膦2 种代谢物之和计。随着牛乳需求量日益增加,为防止草铵膦的滥用,保证牛乳的品质,真正实现乳品行业安全监控,亟需建立牛乳中草铵膦及其代谢物的检测方法。
目前,国内外报道的主要方法有:离子色谱法[10-12]、气相色谱-质谱法[13-15]、毛细管电泳法[16]和液相色谱-三重四极杆串联质谱法[17-28]。离子色谱法存在基质干扰较大,无法有效的分离,灵敏度无法满足限量要求等缺点;气相色谱-质谱法存在前处理繁琐,衍生反应时间长等缺点;毛细管电泳法对于测试液的净化效果要求较高,否则容易堵塞毛细管损坏仪器,同时检测器灵敏度低,不适合复杂基质的痕量分析;液相色谱-三重四极杆串联质谱法在定性和定量上均具有一定的优势,但是仍受限于色谱分离能力和保留能力以及特殊基质的前处理技术。我国尚未发布有关牛乳中草铵膦及其代谢物检测方法的相关标准,国内外鲜有报道,因而急需建立一种实用且快速检测牛乳中草铵膦及其代谢物的分析方法,满足食品安全国家标准的限量规定以及行业的诉求。本研究采用多壁碳纳米管(multi-walled carbon nanotube,MWCNTs)吸附剂作为前处理净化填料,选用一款新型的阴离子农残专用色谱柱,旨在开发一种同时测定牛乳中草铵膦及其2 种代谢物的液相色谱-串联质谱检测方法。图1为草铵膦与2 种代谢物的结构式。

图1 草铵膦及其代谢物的结构式Fig.1 Structural formulas of glufosinate ammonium and its two metabolites
1 材料与方法
1.1 材料与试剂
草铵膦标准品(CAS 号77182-82-2、分子式C5H15N2O4P、纯度≥99%)、3-(甲基膦基)丙酸标准品(CAS号15090-23-0、分子式C4H9O4P、纯度≥99%)N-乙酰基草铵膦标准品(CAS号73634-73-8、分子式C7H14NO5P、纯度≥94%) 美国Dr.Ehrenstorfer公司;草铵膦-D3盐酸盐标准品(CAS号1323254-05-2、分子式C5H10D3N2O4P、纯度≥99%) 美国TRC公司;C18SPE小柱(500 mg,6 mL)、石墨化炭黑(ENVICarb)SPE小柱(500 mg,6 mL) 美国默克公司;PRIME HLB SPE小柱(150 mg,3 mL) 美国Waters公司;MWCNTs(粒径范围10~20 nm,颗粒物长度5~15 μm,比表面积(225±25)m2/g) 天津博纳艾杰尔科技有限公司;PSA和C18填料 美国西格玛公司;一次性针式过滤器(0.2 μm,尼龙),乙腈、甲酸、甲酸铵(色谱级) 上海安谱实验科技股份有限公司;Milli-Q纯水由实验室制备。
1.2 仪器与设备
ACQUITY UPLC TQ-S超高效液相色谱-串联质谱仪、Anionic Polar Pesticide色谱柱(2.1 mm×100 mm,5 μm)、BEH C18色谱柱(100 mm×2.1 mm,1.7 μm)、BEH HILIC色谱柱(100 mm×2.1 mm,1.7 μm) 美国Waters公司;MS 3 basic旋涡振荡器 德国IKA公司;Sorvall Biofuge Stratos高速冷冻离心机 美国Thermo Fisher公司;Millipore Q纯水机 美国Millipore公司。
1.3 方法
1.3.1 标准溶液的配制
草铵膦标准储备液(100 μg/mL):称取草铵膦标准品10.00 mg于100 mL塑料瓶容量瓶中,加入适量的超纯水超声助溶5 min,用水定容至刻度。
草铵膦-D3标准储备液(100 μg/mL):称取草铵膦-D3标准品1.00 mg于10 mL塑料瓶容量瓶中,用超纯水定容至刻度。
N-乙酰基草铵膦标准储备液(100 μg/mL):称取N-乙酰基草铵膦标准品10.00 mg于100 mL塑料瓶容量瓶中,用超纯水定容至刻度。
3-(甲基膦基)丙酸标准储备液(100 μg/mL):称取3-(甲基膦基)丙酸标准品10.00 mg于100 mL塑料瓶容量瓶中,用超纯水定容至刻度。
草铵膦混标中间液(I)(1 μg/mL)的配制:分别量取草铵膦、3-(甲基膦基)丙酸和N-乙酰基草铵膦标准储备液0.25 mL到25 mL塑料容量瓶中,用超纯水定容至刻度。
草铵膦混标中间液(II)(100 ng/mL)的配制:量取草铵膦混标中间液(I)2.5 mL到25 mL塑料容量瓶中,用超纯水定容至刻度。
草铵膦-D3标准中间液(I)(1 μg/mL)的配制:量取草铵膦-D3标准储备液0.25 mL到25 mL塑料容量瓶中,用超纯水定容至刻度。
草铵膦-D3标准中间液(II)(100 ng/mL)的配制:量取草铵膦-D3标准中间液(I)2.5 mL到25 mL塑料容量瓶中,用超纯水定容至刻度。
以上试剂需要存放于聚乙烯或聚丙烯塑料瓶中,0~4 ℃,避光保存。
1.3.2 标准曲线的配制
空白基质标准曲线的配制:选择6 份阴性样品,不加草铵膦-D3标准中间液,其余步骤同1.3.3节样品前处理,制备空白基质溶液。精密量取草铵膦混标中间液(II)(100 ng/mL)0、10、20、50、100、200、500 μL至于不同的进样小瓶中,分别加入草铵膦-D3标准中间液(II)(100 ng/mL)100 μL,分别用空白基质溶液稀释,配制成质量浓度依次为0、1、2、5、1、20、50 ng/mL系列标准工作液,内标质量浓度为10 ng/mL。溶剂标准曲线选用50%甲醇溶液稀释,其余步骤同空白基质标准曲线的配制。
基质效应按下式[29-31]计算:

1.3.3 样品前处理
称取牛乳5 g(精确到0.0001 g),置于30 mL塑料离心管中,加入150 μL草铵膦-D3标准中间液(I)(1 μg/mL)混匀,静置10 min,准确加入10 mL甲醇,涡旋振荡提取10 min,0~4 ℃ 低温冷冻离心(15000 r/min)5 min,取5 mL上清液转移到另外一支离心管中,加入10 mg MWCNTs填料净化,涡旋振荡10 min,0~4 ℃ 低温冷冻离心(15000 r/min)5 min。取1.0 mL上清液于1.5 mL离心管中,0~4 ℃低温冷冻离心(20000 r/min)5 min,取上清液过0.22 μm尼龙针式过滤器于进样小瓶,待测定。
1.3.4 液相色谱-串联质谱条件
1.3.4.1 液相色谱条件
Anionic Polar Pesticide色谱柱(2.1 mm×100 mm,5 μm);柱温50 ℃;进样量10 μL;流动相:0.9%甲酸溶液(A)和0.9%甲酸-乙腈溶液(B);流速0.5 mL/min;线性洗脱梯度:0~3 min,10%~80% A、90%~20% B;3~5 min,80% A、20% B;5~8.0 min,10% A、90% B。
1.3.4.2 质谱条件
电喷雾离子源;数据采集采用负离子扫描;多反应监测模式;脱溶剂温度600 ℃;离子源温度150 ℃,脱溶剂流量1000 L/h;草铵膦及其代谢物的离子对等质谱参数如表1所示。
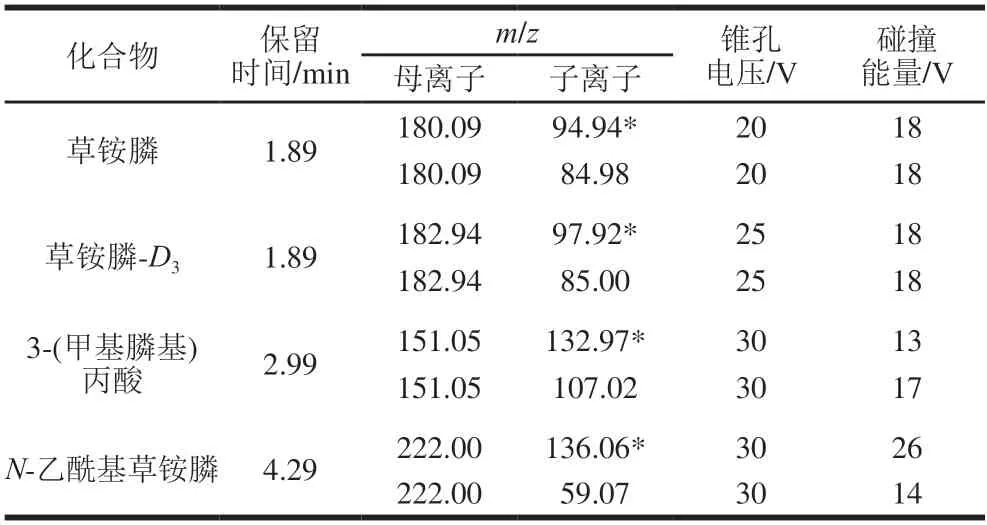
表1 草铵膦及其代谢物质谱参数Table 1 Mass spectrometric parameters of glufosinate ammonium and its metabolites
2 结果与分析
2.1 前处理方法的选择
2.1.1 提取试剂的选择
由于生乳中含有大量的蛋白质,需要去除蛋白,才能进质谱检测,而草铵膦及其代谢物的极性较强,易溶于水、甲醇、乙腈等,同时有机溶剂可以使生乳中的蛋白质变性发生沉淀,因此考察甲醇和乙腈作提取试剂进行分析。结果表明:当选择乙腈作为提取试剂,净化液体中牛乳蛋白质去除的效果较好,但是草铵膦及其代谢物的提取效率不足50%,而当选择加入同等体积的甲醇作为提取试剂,牛乳蛋白质去除效果不如乙腈作为提取试剂,但是提取效率可以达到90%以上,因此选择甲醇作为提取试剂。
2.1.2 净化方法的选择
考察C18SPE小柱(500 mg,6 mL)、石墨化炭黑SPE小柱(500 mg,6 mL)、PRiME HLB SPE小柱(150 mg,3 mL)以及不过SPE小柱净化效果。结果表明,3 种SPE小柱净化后,批次间的重复性较差,重复性均大于20%,不符合方法重复性规定,只有不经过SPE小柱净化的重复性小于10%,需要继续优化。再次考察不过柱直接加入填料的净化效果,分别加入相同质量的C18填料、PSA填料、MWCNTs填料以及不加填料对检测结果的影响。结果表明,草铵膦及其2 种代谢物在3 种填料中净化效果均优于不加填料,其中MWCNTs绝对回收率最高,说明去除基质的能力最佳。MWCNTs是基于多个石墨烯片同轴卷曲结合优秀的纳米技术形成的中空管状立体结构,每层纳米管壁通过sp2与周围3 个碳原子结合,具有较大的比表面积;MWCNTs吸附机理主要集中在π-π作用、疏水作用、H键作用、静电作用力等,与非极性有机化合物和多核芳香化合物相作用,而与极性较强的化合物不会产生吸附作用;牛乳中脂肪等非极性化合物含量较高,样品基质中含有高含量的脂肪会增加质谱电喷雾电离源的表面张力,降低目标物的雾化效率,增强基质效应,影响仪器的灵敏度以及数据稳定性,与文献[32-33]中MWCNTs吸附非极性杂质的效果优于PSA结论一致。因此,选择MWCNTs作为净化填料,见图2。同时,对MWCNTs添加量进行优化,考察5、10、15、20 mg不同添加量,其中5 mg回收率最低,其他不同添加量并无明显差异,因此,选择最小的加入量10 mg。此外。比对净化后的液体不离心和高速低温离心之间的差异。结果表明,净化后的液体不离心较浑浊,不适合精密仪器检测,经高速低温离心后较澄清,可以有效去除油脂以及残留蛋白质等杂质。因此,净化后的液体增加高速低温离心净化步骤。

图2 不同净化填料优化Fig.2 Effect of different column fillers on the recoveries of glufosinate ammonium and its metabolites
2.2 质谱条件优化
草铵膦及其代谢物均属于极性较强的酸性化合物,适合在质谱的负离子模式下进行扫描分析。各目标化合物工作液需要使用50%甲醇溶液稀释,提高目标化合物在仪器上的灵敏度。通过质谱端的蠕动泵注入一定浓度的稀释后标准溶液,在质谱软件的调谐界面优化质谱参数,如电压、温度、气体流速等质谱参数,得到质谱条件,选择2 个响应较强、无干扰的目标物特征离子对,用于定性和定量分析。
2.3 色谱条件优化
2.3.1 色谱柱的选择
考察BEH C18色谱柱(100 mm×2.1 mm,1.7 μm)、BEH HILIC色谱柱(100 mm×2.1 mm,1.7 μm)和Anionic Polar Pesticide色谱柱(2.1 mm×100 mm,5 μm)。结果表明,BEH C18色谱柱(100 mm×2.1 mm,1.7 μm)目标峰保留能力差,不适合草铵膦及其代谢物的分析;BEH HILIC色谱柱(100 mm×2.1 mm,1.7 μm)存在灵敏度低、色谱峰拖尾,且无法实现目标物与基质的有效分离,因此也不适用于草铵膦及其代谢物的分析;Anionic Polar Pesticide色谱柱(2.1 mm×100 mm,5 μm)是在BEH HILIC基础上进行改进,结合WAX(阴离子交换)功能的一款新型的用于分析阴离子农残色谱柱,使该色谱柱对于草铵膦及其代谢物均具有优秀的保留特性和选择性。通过对Anionic Polar Pesticide色谱柱(2.1 mm×100 mm,5 μm)条件的优化,可实现对草铵膦及其代谢物N-乙酰基草铵膦、3-(甲基膦基)丙酸的分离,经过连续数日大量样品的检测,草铵膦及其代谢物N-乙酰基草铵膦、3-(甲基膦基)丙酸出峰时间较稳定,色谱峰较好,受基质干扰小,灵敏度高,满足牛乳中草铵膦及其代谢物检测分析。
2.3.2 液相色谱条件的优化
考察3 种流动相体系,分别为水-乙腈(A)、0.9%甲酸-乙腈(B)、0.9%甲酸-0.9%甲酸乙腈(C)。结果表明,通过对水-乙腈(A)流动相体系优化色谱梯度,目标物的保留能力较差,并未获得较好的色谱峰形;通过对0.9%甲酸-乙腈(B)流动相体系优化色谱梯度,草铵膦色谱峰形得到改善,但是草铵膦的2 个代谢物并未获得较好的色谱峰形;通过对0.9%甲酸-0.9%甲酸乙腈(C)流动相体系优化色谱梯度,草铵膦及其代谢物均获得较好的色谱峰形。此外,对流动相体系中的甲酸含量也进行优化,当体系中甲酸体积分数低于0.9%,均无法获得较好色谱峰形,当系统中甲酸体积分数大于0.9%,目标化合物灵敏度和色谱峰形均未有显著提升,因此选择加入0.9%甲酸。同时对柱温也进行优化,分别考察35、40、45、50、55 ℃,当温度升高到50 ℃,草铵膦及其代谢物峰形均得到改善,并且灵敏度也得到显著提升,当柱温升高到55 ℃,化合物的灵敏度略有下降。因此,柱温定为50 ℃,见图3。

图3 草铵膦及其代谢物离子流色谱图Fig.3 Total ion current chromatograms of glufosinate ammonium and its metabolites
2.4 基质效应
采用标准曲线法实现基质效应的评估分析。标准曲线法优于单点或者多个点单独比较基质效应评价,更具有统计意义和代表性。溶剂标准曲线A:用甲醇溶液(1∶2,V/V)稀释,制成质量浓度分别为0、1、2、5、10、20、50 ng/mL的混合标准工作液;空白基质标准曲线B:用阴性样品得到空白基质溶液,配制成质量浓度与溶剂标准曲线A一致的混合标准工作液。通过空白基质标准曲线B斜率与溶剂标准曲线A斜率比值计算:草铵膦、3-(甲基膦基)丙酸和N-乙酰基草铵膦的基质效应分别为78%、86%和81%。结果表明,3 个化合物的基质效应均为抑制,而草铵膦基质抑制效应略大,为提高草铵膦检测结果的准确性和消除基质效应,草铵膦采用同位素内标法定量,并配制基质标准曲线,对检测结果进行校准。
2.5 标准曲线、检出限和定量限结果
由表2可知,草铵膦及其代谢物添加量为5~20、10~40 μg/kg和10~40 μg/kg时,标准曲线相关系数(R2)均大于0.99,线性相关性良好;检出限为2.5、5.0 μg/kg和5.0 μg/kg(以RSN=3确定检出限),定量限分别为5、10.0 μg/kg和10.0 μg/kg(以RSN=10确定定量限),可以满足分析的需要。

表2 草铵膦及其代谢物的线性范围、线性方程、相关系数、检出限及定量限Table 2 Linear ranges,linear equations,correlation coefficients,and limits of detection and quantification of glufosinate ammonium and its metabolites
2.6 准确度和精密度实验结果
选取空白样品,进行低、中、高3 个水平分别6 个平行进行加标实验,计算回收率和相对标准偏差(relative standard deviation,RSD),结果见表3。研究表明,回收率范围为79.35%~101.80%,RSD为1.15%~8.63%之间,均有良好的准确度和精密度,符合农药残留分析的要求。

表3 加标回收率和RSDTable 3 Recoveries and relative standard deviations for spiked samples
2.7 实际样品测定
利用本研究建立的方法对100 批次不同牧场原奶进行检测,均未检出草铵膦及其代谢物。
3 结论
采用新颖的MWCNTs吸附材料,结合一款新型的阴离子农残专用色谱柱,建立同时检测牛乳中草铵膦及其代谢物残留量检测方法。牛乳中草铵膦及其代谢物3-(甲基膦基)丙酸和N-乙酰基草铵膦的检出限分别为2.5、5.0 μg/kg和5.0 μg/kg,定量限为5.0、10.0 μg/kg和10.0 μg/kg,分别在5、10、20 μg/kg,10、20、40 μg/kg和10、20、40 μg/kg,3 个不同质量浓度水平下的加标回收率范围为79.35%~101.80%,RSD为1.15%~8.63%(n=6);该方法操作便捷、色谱条件稳定、灵敏度高,结果准确,可用于牛乳中草铵膦及其代谢物3-(甲基膦基)丙酸和N-乙酰基草铵膦残留量检测。
猜你喜欢
世界农药(2023年6期)2023-07-05 06:22:48
农村百事通(2021年31期)2021-12-02 01:10:26
食品安全导刊(2021年21期)2021-08-30 08:22:06
农村百事通(2021年11期)2021-01-17 07:33:01
自我保健(2020年8期)2020-01-01 21:12:03
现代园艺(2018年1期)2018-03-15 07:56:12
现代农药(2017年2期)2017-04-18 02:32:44
灾害医学与救援(电子版)(2017年3期)2017-02-06 05:25:10
今日农药(2016年7期)2016-05-14 09:45:30
分析测试学报(2015年9期)2015-12-17 16:44:25
