
慢性子宫内膜炎致炎机制的研究进展
2023-02-04 03:04熊玉晶罗婉彬艾细雄徐艳文
国际生殖健康/计划生育杂志 2023年1期
熊玉晶,罗婉彬,艾细雄,徐艳文
慢性子宫内膜炎(chronic endometritis,CE)是一种局限于子宫内膜的持续炎症性疾病,与不孕、反复妊娠丢失(recurrent pregnancy loss,RPL)和反复种植失败(recurrent implant failure,RIF)等不良妊娠结局密切相关。据文献报道,在不孕、RPL和RIF女性中CE患病率分别为2.8%~56.8%、14.0%~67.5%和9.3%~67.6%[1],且CE可增加流产率和降低单次妊娠活产率[2]。目前CE的诊断主要是通过宫腔镜检查联合免疫组织化学染色观察子宫内膜间质中是否有浆细胞浸润,但尚无统一的诊断标准。其他辅助诊断方法如细菌培养、16S rRNA测序等有其不足之处。此外,经一线治疗(口服多西环素14 d)后,仍有部分患者出现耐药或持续性CE(对2~3个疗程的抗生素治疗不敏感)的情况[1],且研究发现经抗生素治疗后的CE患者体外受精的妊娠结局并未得到改善[3-4]。目前研究认为,CE的形成主要是病原体感染导致子宫内膜炎症调节异常和免疫微环境改变,表现为促炎介质上调、免疫细胞亚群失调及内膜功能异常,但导致CE免疫状态改变和炎症发展的机制尚不明确。本文综述CE可能的发生发展机制,为寻求更规范有效的诊疗措施提供科学依据。
1 炎症通路激活
CE最重要的病原相关模式分子(pathogen associated molecular pattern,PAMP) 是 脂 多 糖(lipopolysaccharide,LPS),还包括来自细菌的脂肽、肽聚糖和病原体的核酸等。研究发现,损伤或死亡细胞释放的染色质DNA或含有自身抗原的免疫复合物是重要的刺激因子[5]。Chen等[6]对32例CE患者和72例非CE患者的子宫内膜进行转录组学分析发现,差异基因的功能主要富集于免疫和炎症相关生物过程,且在富集的通路网络图中,细胞因子受体相互作用通路和Toll样受体(Toll-like receptor,TLR)信号通路与其他通路存在相互联系。此外,受到LPS刺激的牛子宫内膜上皮细胞(bovine endometrial epithelial cell,BEEC)的转录组学分析提示,LPS可调控BEEC的炎症和免疫反应,并显著上调与炎症相关通路的表达,包括NOD样受体(NOD-like receptor,NLR)信号通路、TLR信号通路、细胞因子受体相互作用通路、趋化因子信号通路、抗原加工和提呈通路[7]。
在CE患者子宫内膜和经LPS刺激后的子宫内膜上皮细胞以及间质细胞中均发现促炎介质和趋化因子表达显著上调[8]。以上研究提示,LPS等PAMP可作用于TLR、NLR等模式识别受体激活炎症通路,通过启动一系列信号转导和级联反应显著上调促炎因子和趋化因子表达,并促进子宫内膜免疫紊乱和炎症微环境形成。
1.1 TLR通路异常激活TLR是一类能识别PAMP的受体,每种TLR在PAMP识别和免疫反应方面都有不同的功能。其中TLR4是识别细菌LPS的关键受体,TLR2则参与来自多种微生物的多种PAMP识别,两者与CE的发生关系密切。此外,损伤或死亡细胞释放的内源性分子,如高迁移率族蛋白B1(high mobility group protein 1,HMGB1)和热休克蛋白,可被TLR2、TLR4或TLR2-TLR4异 二 聚 体 识 别[5]。HMGB1与病原体或自身DNA结合形成复合物后,还可通过抗原提呈细胞的晚期糖基化终产物受体内化而与胞质内的TLR7和TLR9结合[9]。由此可见,微生物感染可能会诱发修饰性内源性分子释放,这些内源性分子与PAMP被TLR或其他胞质内模式识别受体识别后可诱发炎症。因此,继发性局部自身免疫反应可能是持续性CE患者经抗生素治疗清除病原体后仍持续存在子宫内膜炎症表现的重要原因。
TLR通路的过度激活参与CE的发生。TLR与PAMP结合后可通过髓样分化因子88(myeloid differentiation factor 88,MyD88)依赖或非依赖性途径激活其下游的核因子κB(nuclear factor kappa beta,NF-κB)和(或)丝裂原活化蛋白激酶(mitogenactivated protein kinase,MAPK)信号通路,促进各种促炎介质的转录,导致大量炎性细胞浸润。多项体外细胞实验证实,NF-κB激活可导致子宫内膜炎,淫羊藿苷可通过抑制TLR4/NF-κB通路激活核因子E2相关因子2(nuclear factor E2-related factor 2,Nrf2)通路来改善氧化应激,抑制LPS诱导的小鼠子宫内膜炎[10]。此外,TLR通路的负调控因子的突变或表达下调可能导致体内持续发生炎症反应。如蛋白激酶B1(protein kinase B1,PKB1或Akt1)可抑制TLR诱导的MyD88磷酸化,在敲除Akt1基因的RAW264.7细胞中,LPS可增强NF-κB和β干扰素的启动子活性,并诱导炎症因子表达上调[11]。
1.2 NLR通路异常激活NLR可以识别胞质中的PAMP和生物分子,在免疫细胞和成纤维细胞、上皮细胞等非免疫细胞中均有表达。其中,NOD样受体蛋白1(NOD-like receptor protein 1,NLRP1)和NLRP2识别细菌细胞壁成分的降解产物,NLRP3响应各种刺激形成炎性小体复合体,并通过活化胱天蛋白酶1(caspase-1)促进白细胞介素1β(interleukin-1β,IL-1β)和IL-18的释放。既往研究表明,炎症是子宫内膜异位症、多囊卵巢综合征及RPL等多种生殖相关疾病的重要病理生理基础。多囊卵巢综合征小鼠的纤维化卵巢组织、特发性RPL患者的子宫内膜组织中均有NLRP3炎性小体激活[12],提示NLRP3炎性小体可能参与慢性炎症过程。目前,有数个研究提示NLRP3炎性小体与子宫内膜炎症反应有关。体外实验显示,用LPS刺激牛原代子宫内膜上皮细胞、间质细胞及外周血单核细胞后均可促进IL-1β分泌,并以间质成纤维细胞反应最显著[13]。NLRP3受体抑制剂或caspase-4特异性小干扰RNA(small interfering RNA,siRNA)则能阻断子宫内膜细胞IL-1β的产生。一项小鼠CE模型研究证实,LPS通过诱导内质网应激激活硫氧还蛋白相互作用蛋白(thioredoxin interacting protein,TXNIP,可激活NLRP3通路)促进IL-1β表达[14]。经LPS刺激后,山羊子宫内膜基质细胞中内质网应激标志蛋白、自噬标志物和炎症因子表达水平上调,应用内质网应激抑制剂4-苯基丁酸后,上述现象消失[15]。但目前尚无关于CE患者子宫内膜中NLRP3炎性小体激活情况的报道。
多种机制参与NLR通路调控。除内质网应激可激活NLRP3通路外[14-15],在氧化应激和炎症等病理条件下,TLR通路可上调NLRP3、IL-1β前体(pro-IL-1β)和pro-IL-18的表达,当NLRP3表达量达到一定水平时即可激活NLRP3炎性小体[16]。体外细胞实验显示,将HMGB1和绒毛滋养细胞共孵育后可上调NLRP3表达[17],提示HMGB1亦能激活NLR通路并诱导胚胎的炎症及损伤。另有研究发现,上皮细胞外升高的ATP可激活子宫内膜的巨噬细胞NLRP3炎性小体,提示NLRP3炎性小体可能参与无菌性炎症的发生[18]。目前关于CE中NLR通路如何“启动”、调控并最终导致CE形成仍不十分清楚。Lu等[19]报道激活的NLRP3炎性小体可调节辅助性T细胞17(T helper cell 17,Th17细胞)和调节性T细胞(regulatory T cell,Treg细胞)的平衡,参与RPL的发病过程。而部分CE患者内膜亦存在Th17/Treg细胞失衡[6,20],因此推测,NLRP3可能通过改变子宫内膜免疫状态参与CE形成。
综上,CE炎症反应的发生发展是以TLR和NLR通路为中心的多条炎症通路相互作用的结果,但各通路间的联系尚不清楚。充分了解各通路间的相互关系,寻找到网络间关联的关键分子,将为CE的治疗提供新的思路。
2 子宫内膜免疫细胞代谢改变
研究发现,CE子宫内膜中免疫细胞亚群发生改变,主要表现为效应T细胞、M1型巨噬细胞等促炎型细胞增加[20]。免疫细胞的表型稳定及功能行使需要相应的代谢状态,因此免疫细胞代谢改变可能是子宫内膜免疫紊乱与炎症微环境形成的重要机制。
哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是哺乳动物重要的代谢传感器,可参与调控细胞代谢,如激活的mTOR信号通路可增强缺氧诱导因子1α介导的葡萄糖摄取和糖酵解,促进CD4+T细胞活化和Th17/Th1细胞分化[21]。糖酵解增强倾向于支持效应T细胞、M1型巨噬细胞等细胞的促炎功能以及各免疫细胞的增殖、迁移行为,并可抑制叉头蛋白3(forkhead box protein 3,FOXP3)表达和降低Treg细胞表型的稳定性[22],从而有利于发挥促炎效应。研究发现PAMP可激活TLR、T细胞受体等相关受体,调单核巨噬细胞的mTOR信号通路[23]。相反,转化生长因子β(transforming growth factor-beta,TGF-β)信号通路、IL-4信号通路则通过抑制糖酵解、促进线粒体氧化和脂肪酸氧化支持Treg细胞、M2型巨噬细胞等的抗炎功能[24],CE中TGF-β、IL-4等抑炎因子减少可能进一步增强糖酵解和炎症反应。
此外,脂质合成代谢对免疫细胞的影响也逐渐被研究者所关注。最近的研究发现,经LPS刺激的人原代巨噬细胞中,在炎症反应后期固醇调节元件结合蛋白1(sterol regulatory element-binding protein 1,SREBP1)可介导脂质代谢重编程促进炎症消散,即SREBP1通过激活参与不饱和脂肪酸生物合成的基因,促进鞘脂从头合成,从而抑制TLR4/NF-κB通路调控基因的表达[25]。但CE子宫内膜中免疫细胞的脂质合成代谢变化及在CE形成中的作用尚不清楚。因此CE中过度激活的炎症通路和促炎介质可能通过改变免疫细胞代谢调控免疫细胞增殖、分化和功能状态,最终造成子宫内膜免疫紊乱与炎症微环境形成。
3 微小RNA(microRNA,miRNA)介导炎症发生发展
3.1调控CE炎症通路的miRNAmiRNA是一类长度为20~22个核苷酸的非编码单链RNA分子,主要通过结合到特定靶mRNA的3'端非翻译区域抑制其翻译而发挥转录后调控作用。Lv等[26]研究显示,LPS刺激牛子宫内膜间质细胞(bovine endometrial stromal cell,BESC)后,差异表达的miRNA显著富集于MAPK、肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)和IL-17信号通路,提示miRNA参与LPS的炎症致病机制。miRNA可通过调控相关炎症通路的关键分子参与CE的发生发展,具体调节靶点见表1[8,26-33]。实际上,miRNA与mRNA存在复杂的调控网络,而目前关于miRNA与CE的研究多仅在单一种类细胞层面进行,将来仍需在子宫类器官或人类子宫内膜组织中验证细胞层面失调的miRNA在CE中的实际作用。
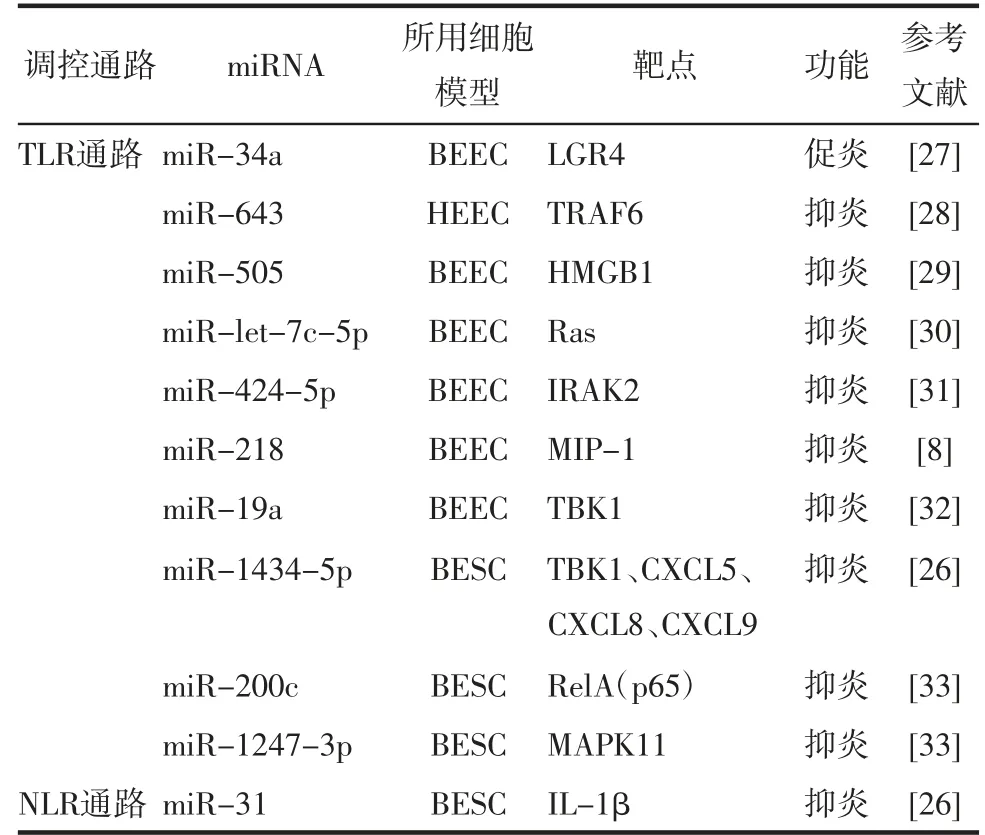
表1 调控CE炎症通路的miRNA
此外,目前的研究多集中在miRNA对子宫内膜上皮和间质细胞的炎症通路的调控作用,关于miRNA对免疫细胞影响的研究有限。Lv等[26]的研究提示,经LPS刺激的BESC中差异表达的miRNA显著富集于细胞代谢过程,但目前尚无报道关于miRNA如何调控细胞代谢促进CE发生的相关机制研究。鉴于miRNA在炎症、细胞发育和增殖等多个生理病理过程中的重要作用,仍有必要进一步研究miRNA在CE中的作用。
3.2 外泌体源性miRNA调控子宫内膜炎症反应外泌体可由上皮细胞、间质细胞、大多数免疫细胞等宿主细胞和微生物分泌到细胞外间隙,并将蛋白质、脂质、mRNA或miRNA等物质转移到靶细胞,调节靶细胞功能[34]。如Treg细胞释放外泌体,并将外泌体源性miR-let-7d转移到Th1细胞,抑制后者增殖和γ干扰素分泌,从而抑制炎症反应[35]。宫腔液外泌体源性miRNA可在调控CE炎症反应中发挥重要作用。研究发现,患子宫内膜炎的牛宫腔液外泌体miRNA发生失调,其中BEEC分泌的外泌体源性miR-218减少,导致抑制靶细胞MIP-1表达的作用减弱而使炎症发展[8]。此外,外泌体还可通过传递PAMP等抗原物质发挥显著的免疫调节活性,参与炎症的调控。因此,外泌体可作为重要的细胞间通讯媒介在CE子宫内膜免疫状态紊乱和炎症反应的发生中发挥重要调控作用。
4 DNA甲基化模式异常
微生物感染可诱导宿主细胞DNA去甲基化。研究发现,LPS可改变BEEC的DNA甲基化模式,大部分差异基因表现为低甲基化与基因表达增加,其中差异甲基化基因主要是与免疫功能、炎症过程、细胞增殖、凋亡、细胞黏附及细胞外基质重塑等相关的蛋白编码基因[36]。LPS所致子宫内膜炎症可能与上述基因的甲基化变化有关,如LPS诱导BEEC的AKT1基因甲基化和IL-1受体相关激酶1(IL-1 receptor-associated kinase 1,IRAK1)基因低甲基化,进而激活TLR/NF-κB通路[36]。此外,LPS促进IL-6和IL-8启动子区域发生DNA去甲基化,且与IL-6、IL-8表达量增加一致[36]。Ⅱ类组蛋白脱乙酰酶(histone deacetylase,HDAC)基因低甲基化上调HDAC表达,可能通过多种途径促进炎症发生发展。如HDAC可调节淋巴细胞发育和功能相关的信号通路、维持缺氧诱导因子1α的稳定性和转录活性、调控TLR通路关键分子的乙酰化水平和活性[37]。鉴于LPS可引起大量炎症相关基因的甲基化改变,推测微生物感染可能会诱发基因调控区域DNA甲基化变化和基因转录的持续异常,进而导致CE患者子宫内膜炎症长期存在。
目前CE患者子宫内膜DNA甲基化水平和DNA甲基化模式的改变及其对子宫内膜功能和炎症反应发生的影响仍不清楚。多项研究证实,人类子宫内膜不同周期阶段的DNA甲基化水平是动态变化的,且子宫内膜异位症、子宫内膜癌等内膜疾病有其特异的DNA甲基化模式[38],因此未来可通过单细胞测序技术以获得CE子宫内膜周期特定阶段特定细胞类型的DNA甲基化谱,以全面评估CE子宫内膜DNA甲基化与子宫内膜炎症、内膜容受性之间的关系,有助于发现更精确的生物标志物和新的潜在治疗靶点。
5 结语
综上所述,病原体感染等刺激因素可激活以TLR通路、NLR通路为核心的炎症通路网络,进而导致大量炎性介质释放。炎症通路的激活还可通过以调控mTOR通路为代表的途径改变子宫内膜免疫细胞的代谢活动。上述改变最终导致子宫内膜免疫紊乱与炎症微环境形成。miRNA失调、DNA甲基化模式异常等机制可通过异常调控相关炎症通路或干扰炎性介质生成参与CE的发生发展。鉴于目前CE的诊疗方法的规范性和有效性尚欠缺,对CE的致炎机制进行深入探索有助于寻找关键的诊疗靶点和改善妊娠结局。如通过宫腔内给予外源性外泌体以补充下调的miRNA,或应用雷帕霉素抑制过度激活的mTOR通路等可能有助于更好地治疗CE。此外,与CE炎症相关的各种异常分子或病理生理过程对子宫内膜容受性和妊娠生理的影响机制尚不十分清楚,仍有待进一步研究。
猜你喜欢
中华养生保健(2020年9期)2021-01-18
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
中外医疗(2016年15期)2016-12-01
作文教学研究(2016年1期)2016-07-05
中华老年多器官疾病杂志(2016年9期)2016-04-28
医学研究杂志(2015年8期)2015-06-22
现代检验医学杂志(2015年2期)2015-02-06
沈阳医学院学报(2014年4期)2014-12-27
中国中医药现代远程教育(2014年15期)2014-03-01
