
Ligand-field regulated superalkali behavior of the aluminum-based clusters with distinct shell occupancy
2023-01-30 06:48JunLiMingweiCuiHunYngJingChenShioCheng
Chinese Chemical Letters 2022年12期
Jun Li ,Mingwei Cui ,Hun Yng ,Jing Chen,Shio Cheng ,*
a School of Chemistry and Chemical Engineering,Shandong University,Ji’nan 250100,China
b School of Physics,Shandong University,Ji’nan 250100,China
Keywords:Superalkali Density functional theory Ligand-field strategy Aluminum-based cluster Electronic property
ABSTRACT Protecting clusters from coalescing by ligands has been universally adopted in the chemical synthesis of atomically precise clusters.Apart from the stabilization role,the effect of ligands on the electronic properties of cluster cores in constructing superatoms,however,has not been well understood.In this letter,a comprehensive theoretical study about the effect of an organic ligand,methylated N-heterocyclic carbene(C5N2H8),on the geometrical and electronic properties of the aluminum-based clusters XAl12(X=Al,C and P)featuring different valence electron shells was conducted by utilizing the density functional theory(DFT)calculations.It was observed that the ligand can dramatically alter the electronic properties of these aluminum-based clusters while maintaining their structural stability.More intriguingly,different from classical superatom design strategies,the proposed ligation strategy was evidenced to possess the capability of remarkably reducing the ionization potentials(IP)of these clusters forming the ligated superalkalis,which is regardless of their shell occupancy.The charge transfer complex formed during the ligation process,which regulates the electronic spectrum through the electrostatic Coulomb potential,was suggested to be responsible for such an IP drop.The ligation strategy highlighted here may provide promising opportunities in realizing the superatom synthesis in the liquid phase.
Due to the quantum confinement,atomic clusters,containing several to thousands of atoms,usually present unique properties,which represents a new phase of materials bridging the gap between the atoms and bulk materials.It is acknowledged that the composition and structure both have significant impacts on the cluster’s properties[1,2].Particularly,a class of atomic clusters has been discovered to have the characteristics in mimicking the chemistry of the atom in the periodic table,termed as“superatoms”[3–5].Since they share similar properties with atoms and expand the definition of an element to involve species that behave as entities,the superatoms could be utilized to build a threedimensional(3D)periodic table[6].Serving as building blocks,these superatoms are also potential candidates in forming novel cluster-assembled materials,which can be used in the energy storage and conversion[7–12].
In the development of the superatoms,different categories have been proposed,such as superalkali[10,13],superhalogen[12,14,15],rare-earth superatoms[16]and magnetic superatoms[17,18].Among them,the superalkali with the adiabatic ionization potentials(AIPs)lower than those of alkali atoms(3.89-5.39 eV)and superhalogen,whose adiabatic electron affinities(AEAs)are higher than those of halogen elements(3.06-3.62 eV),are two most important series that could mimic the properties of alkali and halogen atoms,respectively.Over the past several decades,various electron counting rules,such as the Jellium model[19],the octet rule[20,21],the Wade-Mingos rule[22,23],the 18-electron rule[24],the aromatic rule[25,26]and others,have been successfully applied in constructing superatomic clusters.Yet,challenges remain in designing and synthesizing these special clusters since these classical models highly correlate with the intrinsic properties of clusters,e.g.,the elemental composition and the shell filling,which are relatively difficult to modulate in the chemical synthesis.In other words,conventional design strategies mainly depend on how the superatomic states of the superatoms are filled.Under such circumstance,an interesting question is whether one can control the electronic properties of clusters by only regulating their external environment rather than altering their intrinsic characteristics?That is to say,is it possible to go beyond the traditional shell effect to manipulate the clusters’electronic properties,for example,to turn the superhalogen into a superalkali,or transform the clusters featuring closed shell which obeys the Jellium model into a superalkali one?This represents a substantial step towards the chemical synthesis of atomically precise superatoms.Fortunately,it is well-established that coating clusters with ligands is a practical way that has been proved by numerous experiments[27–31],where ligands were used to stabilize the cluster cores.Our previous works have also proved that it is effective to tailor the electronic properties of metal clusters by applying suitable ligands[10,32,33].
In the present letter,we combined the ligation strategy,which is wildly used in the liquid-phase cluster synthesis,with the bare Al13and doped XAl12(X=C and P)clusters to examine the ligandfield effect on the electronic properties of these clusters.Considering the limitations of traditional electron counting rules,which are inseparable from the shell effect of the cluster itself or its unique geometric configuration,what we interest most here is whether it is attainable to transcend the shell effect and produce superalkalis without changing the cluster’s electronic configuration and stability.Our long-term experience in designing superatoms[10,12,16,32-35]and the achievements of our peers[36–38]inspire us that it may achieve the intended purpose by connecting the electron-donating ligand to the cluster core.Thus,one class of typical ligands,N-heterocyclic carbenes(NHCs)that have been successfully applied in the organic chemistry and coordination chemistry,was adopted here.Not only can NHCs serve as the reagents and catalysts in organic chemistry,such as the transesterification,nucleophilic aromatic substitution,and cycloaddition reaction,but they also play an important role in the coordination chemistry and organometallic reaction[39–41].To simplify the calculations,we chose the less“bulky”one,1,3-dimethylimidazol-2-ylidene(C5N2H8),a neutral compound containing a divalent carbon atom with an incomplete electron octet.Successive attachment of C5N2H8on these aluminum-based clusters featuring different valence electrons,i.e.,39,40 and 41e,was attempted to examine its effect on the clusters’geometrical and electronic properties.Furthermore,the mechanism for the change in the electronic property and the interaction between the ligand orbital and superatomic states of these clusters were also discussed.
All calculations were performed by using the Gaussian 09 program package[42].The PBE0[43]functional coupled with the def2-SVP basis set[44]was employed to obtain the optimized structures of XAl12(C5N2H8)n(X=Al,C and P;n=1-3).Various positions of ligands and spin structures were checked and compared to ensure the ground states of these clusters,and no imaginary frequencies were observed.To evaluate the AIP and AEA values,geometries of all the neutral,cationic,and anionic clusters were optimized.Our calculated EA of Al13is 3.54 eV.This result is in excellent agreement with the experimental measurement(3.57±0.05 eV)[45],validating the accuracy of the present level of theory.The Hirshfeld,Mulliken and NBO charge were analyzed to estimate the amount of charge transfer between the ligand and the metallic core in the ligated aluminum-based clusters[46,47].The electrostatic potential(ESP)analysis,molecular orbital(MO)analysis,and charge decomposition analysis(CDA)were also conducted and visualized via the Multiwfn[48]and VMD[49]softwares.
Considering numerous possible configurations and spin multiplicities,we first optimized the ground states of XAl12(C5N2H8)n(X=Al,C and P;n=1-3)clusters at the PBE0/def2-SVP level.Their neutral and charged ground-state structures are depicted in Fig.1 and Figs.S1 and S2 (Supporting information),respectively.Here,the XAl12clusters featuring 39,40 and 41 valence electrons,respectively,were adopted as the cluster cores since we want to explore whether the effect of C5N2H8in regulating the clusters’electronic properties is independent of their shell fillings.The 40-electron clusters,i.e.,CAl12and Al13−,share the identical Ihsymmetry,whose shell configurations are(1S)2(1P)6(1D)10(2S)2(1F)14(2P)6based on the Jellium model.And,once the center atom of CAl12is replaced by Al or P,the electronic shell rearranges to…(1D)10(2S)2(2P)6(1F)13or…(1D)10(2S)2(1F)14(2P)6(1G)1,respectively.As shown in Fig.1,there is a significant change in the bond lengths of the doped clusters,representing a symmetry lowering in these clusters compared with the original CAl12cluster.Specifically,compared with CAl12,the average bond lengths from the central atom to the surface Al atoms of Al13and PAl12increase from 2.53˚A to 2.67 and 2.65˚A.Such a change in geometries can be attributed to the Jahn-Teller distortion.
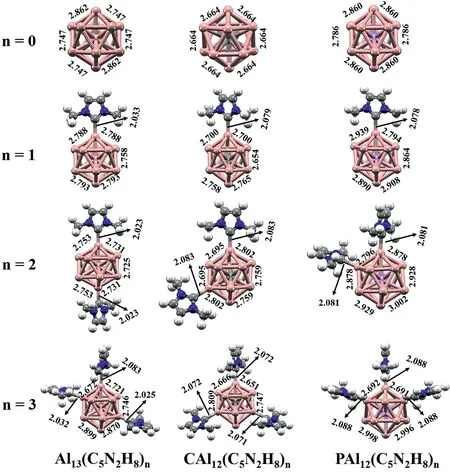
Fig.1.Optimized ground states of XAl12(C5N2H8)n(X=Al,C and P;n=1-3)calculated at the PBE0/def2-SVP level of theory.The Al-Al and Al-C bond lengths are shown in˚A.
The structures and relative stability of the ligated complexes were also investigated.As shown in Fig.1,the structures are quite different when the 2ndC5N2H8ligand was connected to XAl12(X=Al,C and P).In Al13(C5N2H8)2,the 2ndligand occupies the para-position,while the two ligands are in the orthoand meta-positions of PAl12(C5N2H8)2and CAl12(C5N2H8)2,respectively.Furthermore,when these clusters bind with the 3rdligand,Al13(C5N2H8)3stabilizes in an ortho-position structure,while the other two tendencies remain unchanged.With the continuous attachment of the ligand,the symmetry of the cluster gradually decreases.Surprisingly,although the symmetrical distortion takes place during the ligation,the average bond lengths between the central atoms X and the surficial Al atoms in different ligated clusters scarcely change(Fig.2a).Thus,it seems that the geometry of XAl12remains almost unchanged after interacting with C5N2H8.Does this imply that the bonding characteristics and electronic arrangements of the cluster cores have not changed significantly during the ligation process?
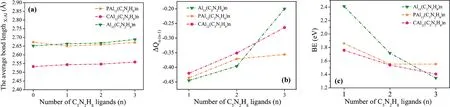
Fig.2.Calculated(a)average bond lengths between the central atom X to surface Al atoms,(b)accumulated charges of the inner-core clusters,and(c)BEs in XAl12(C5N2H8)n(X=Al,C and P;n=1-3).
To gain the charge transfer behavior between the inner-core and ligands,the Hirshfeld,Mulliken and NBO charge analysis were performed.As summarized in Table S1(Supporting information),consistent trends and conclusions were reached.Taking the Hirshfeld charge analysis as an example,ΔQn-(n-1)represents the charge accumulation on the inner-core with respect to its precursor ligation.It can be seen from Fig.2b that the charges of all inner cores increase monotonously with the continuous addition of C5N2H8.To understand the overall charge transfer behavior,the total charge transfer amountΔQn,which represents the charge accumulation on the inner core with respect to its bare cluster,was also examined.Based on the present calculations,the total charge transfers to the Al13,CAl12and PAl12cluster cores are 1.05,1.17 and 1.03e upon the attachment of three C5N2H8ligands,respectively,which clearly demonstrates the electron-donating characteristic of C5N2H8.To visualize such charge distribution,the ESP of XAl12(C5N2H8)n(X=Al,C and P;n=1-3)were drawn.As shown in Fig.S3(Supporting information),the surface of the naked cluster is in dark blue,signifying the negative charge accumulation,and the color continues to deepen as the number of C5N2H8increases,which is the same as the results of the charge analysis(Fig.2b).
To further explore the interactions between C5N2H8and three aluminum-based clusters,the binding energies(BEs)(Fig.2c)were calculated as:
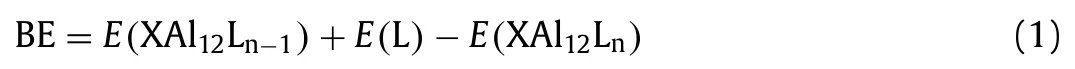
where L=C5N2H8,andEpresents the total energies of different species.Due to the electron deficiency of Al13,the BE of the 1stligand attachment to Al13is much larger than those of other two clusters.The calculated BEs are in the range of 1.35-2.41 eV,which are higher than the previous reported PH3and CO ligands(0.29-1.17 eV)[10].This indicates that the interactions between C5N2H8and XAl12(X=Al,C and P)are stronger than those of the CO and PH3ligands,which may increase the probability of being obtained in the liquid-phase cluster synthesis.In addition,the energy gap between the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO),marked as H-L gap,is a useful criterion to assess the relative stability of clusters.Generally,clusters with larger H-L gap are more stable and chemically inert[50–52].The calculated H-L gaps of XAl12(C5N2H8)n(X=Al,C and P;n=1-3)are in the range of 1.44-2.84 eV(Table S2 in Supporting information).These high H-L gaps are comparable with that of C60(1.70 eV)[53],demonstrating the high stability of these ligated clusters.
We now turn our attention in clarifying whether such strong ligation process can alert the electronic properties of these clusters since all calculated BEs are relatively large(Fig.2c).Here,two important electronic properties,i.e.,AIP and AEA,of ligated clusters were estimated,which are defined as:

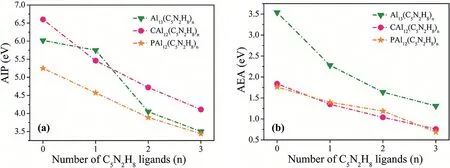
Fig.3.Calculated AEAs and AIPs of the ligated XAl12(C5N2H8)n complexes(X=Al,C and P;n=0-3).All values are given in eV.
The theoretical AEA and AIP values are listed in Table S3(Supporting information).As mentioned above,Al13(39e)lacks an electron to satisfy the closed shell.Therefore,Al13is expected to exhibit superatomic character analogous to halogen atoms.Our calculated AEA value of Al13is 3.54 eV,which is similar to that of the halogen.On the contrary,PAl12(41e)is easy to lose an electron,showing the property of the superalkali metal(AIP=5.25 eV),which is consistent with the experimental data[36,46].The CAl12cluster(40e)exhibits superior stability due to its electronic closedshell structure with lower EA and higher AIP values.Such phenomenon can be illustrated by their electronic structures according to the Jellium model.
The AEAs and AIPs of XAl12(C5N2H8)nwere computed to evaluate the electronic regulation ability of C5N2H8.As a result,a successive decline in their AIPs and AEAs with the continuous addition of C5N2H8was observed(Fig.3).Particularly,the Al13superhalogen cluster changed to a superalkali once the 2ndC5N2H8was attached to the naked cluster.Similar trends were also found in another two clusters,CAl12and PAl12.As shown in Table S3(Supporting information),the AIP of the closed-shell CAl12cluster reduced from the original 6.60 eV to 4.72 eV in CAl12(C5N2H8)2,and that of the superalkali PAl12cluster continues to decrease to 3.89 eV(PAl12(C5N2H8)2).This finding is interesting because only two C5N2H8ligands can strikingly decrease the AIPs of these aluminum-based superatomic clusters with different shell structures,i.e.,39,40 and 41 e.Particularly,the Al13and CAl12clusters transform into superalkalis upon the attachment of only two C5N2H8ligands.Additionally,the AIPs of Al13(C5N2H8)3and PAl12(C5N2H8)3go further down to 3.50 and 3.44 eV,respectively,which are lower than those of any alkali atoms in the periodic table.These results undoubtedly show that,no matter how their electronic shells are filled(closed or not),C5N2H8exhibits strong power in modulating the electronic properties of these clusters.The conversion between the aluminum-based superatomic clusters and superalkali metals can be attributed to the strong electronic regulation ability of C5N2H8,which is not associated with the filling of the electronic shell.
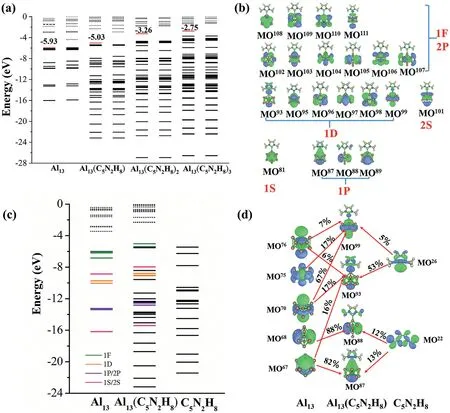
Fig.4.(a)One-electron energy levels of Al13(C5N2H8)n(n=0–3).The red lines indicate the MOs where the ionization occurs;(b)maps of theαoccupied MOs of Al13(C5N2H8),where superscripts indicate the orbital numbers;(c)CDA analysis of Al13(C5N2H8)with Al13 and C5N2H8 as fragments,and their angular characteristics are marked with different colors;(d)isosurface graphs of partial orbital interaction diagram.The numbers marked beside the red lines indicate the contribution from the fragmental MO to the MOs of Al13(C5N2H8).The occupied and unoccupied states in(a)and(c)are illustrated by solid and dashed lines,and the isosurface graphs in(b)and(d)are plotted with an isosurface value of 0.03.
To further understand the origin of the decrease in AIPs of these ligated clusters,we examined their one-electron energy levels(Fig.4a,Figs.S4a and S5a in Supporting information).It can be seen from Fig.4a that the continuous increment of the number of the ligand significantly lifts the energy levels of the HOMOs of Al13(C5N2H8)n(n=0-3)with the values varying from−5.93 eV to−2.75 eV.It is well-accepted that the ionization process usually occurs in the HOMO of the molecule.Thus,this finding corresponds to the reduction of the theoretical AIPs of Al13(C5N2H8)n(n=0-3),which decreases from the original 6.02 eV to 3.50 eV.Similar situations also exist in CAl12(C5N2H8)nand PAl12(C5N2H8)n(n=0-3)(Figs.S4a and S5a).Therefore,it is reasonable to suggest that the prominent decline of the AIPs of XAl12(C5N2H8)n(X=Al,C and P;n=0-3),leading to the formation of superalkali clusters,stems from the upward shift of the electronic spectrum.
It is worth noting that,although the successive ligation lifts the energy levels of the metallic core,the order of energy levels in different superatomic MOs almost stays unchanged compared with the bare one.More interestingly,there are no decided change in the H-L gaps during the ligation process(Table S2 in Supporting information),implying that the electronic structures or the shell fillings of the cluster cores may have not been damaged.To verify this conjecture,the MOs of XAl12(C5N2H8)(X=Al,C and P)complexes were carefully checked.Strikingly,all superatomic MOs of these three aluminum-based cluster cores are well preserved and identified.For example,as shown in Fig.4b,the superatomic MOs of Al13(C5N2H8)are obvious,which is(1S)2(1P)6(1D)10(2S)2(2P)6(1F)13.Similar situations also exist in CAl12(C5N2H8)and PAl12(C5N2H8),as evidenced in Figs.S4b and S5b(Supporting information).
To gain a deeper understanding about the nature of the interaction between the Al13superatomic states and C5N2H8,the charge decomposition analysis(CDA),a method based on the concept of fragment orbital to decompose the charge transfer between molecular fragments into the contribution of orbitals,was conducted to evaluate the charge transfer between these two moieties.Accounting to the CDA analysis(Figs.4c and d),the electron transfer from the ligand to Al13is 0.487e in Al13(C5N2H8),which is consistent with the previous Mulliken analysis.As displayed in Fig.4c,the frontier 1F and 2P superatomic MOs of the ligated complexes near HOMO have almost no interaction with C5N2H8.The energy levels of Al13near the HOMO move upward with the connection of C5N2H8.It is prominent that the occupied MO93of Al13(C5N2H8)is responsible for the interactions between Al13and C5N2H8,as shown in Fig.4d.This MO,which is mainly formed by the HOMO(MO26,53%)of C5N2H8and MO67(16%),MO70(17%),and MO76(6%)of Al13,exhibits the largest electron transfer compared with all other MOs.The MO67(82%)and MO68(88%)of Al13display similar electron characteristic and intermingle with the MO22of C5N2H8leading to the formation of the occupied MO87and MO88of Al13(C5N2H8),respectively.More CDA information and partial orbital interaction diagrams for CAl12(C5N2H8)and PAl12(C5N2H8)are given in Figs.S4c and d,S5c and d(Supporting information),respectively.
Obviously,above findings clearly demonstrate that the electrondonating C5N2H8ligand possesses the power in dramatically dropping the AIPs of typical aluminum-based clusters featuring different shell configurations without disturbing their spueratomic states.Such regulation effect induced by the ligand-field,which is independent of the filling of the electronic shell of the cluster,is completely different from traditional superalkali construction strategies.Importantly,considering the huge application of the ligands in the chemical synthesis of clusters,the present ligand-field strategy cannot only modulate the electronic properties of clusters leading to the formation of novel superalkalis,but also provides promising potential in synthesizing ligated superalklis in the liquid phase.Superior to conventional superatom design strategies that mainly alter the electronic shell filling of the clusters,the current ligand-field strategy has potential to broaden the practical applications of the superatoms.In addition,the C5N2H8ligand was just adopted as a model ligand here,and other ligands featuring similar electron-donating characteristic may also play a part in constructing such superalkali.
In conclusion,the ligand-field strategy,unlike traditional electron counting rules,has been investigated systematically by employing the DFT calculations on the aluminum-based clusters with different shell configurations.During the C5N2H8ligation process,the icosahedral geometries of the metallic moieties,XAl12(X=Al,C and P),remain almost unchanged,indicating that the ligand did not remarkably influence the clusters’structural stability.Relative energetic stability and electronic characteristics of these clusters were also examined.It was observed that the C5N2H8ligand can significantly reduce the AIPs of these aluminum-based clusters without altering their superatomic states.This phenomenon is intriguing because it demonstrates that the regulation effect of such ligand on the clusters’electronic property is irrespective of the filling of the shell configuration of the cluster,which is apparently different from traditional superatom design strategies.The result of the AIP drop may be ascribed to the charge transfer complexes formed by C5N2H8,which increases the energy of the HOMO level through the electrostatic Coulomb potential.Moreover,the ligand,rather than just protecting the metallic core from aggregating,can regulate the electronic property of clusters to construct multiple superalkali species.These findings provide potential opportunities for the superatom synthesis in the liquid phase since ligands are widely utilized in the chemical synthesis of atomically precise clusters,which will broaden the practical applications of the clusterassembly materials.We hope this study will spur further studies about the novel properties and phenomenon of sub-nanoclusters,especially for mimicking the rare/noble elements by using the clusters composed of earth-abundant elements,both in the theory and experiments.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work is supported by the Innovation Project of Jinan Science and Technology Bureau(No.2021GXRC032),the Taishan Scholars Project of Shandong Province(No.ts201712011),the National Natural Science Foundation of China(NSFC,Nos.92161101,21603119),the Natural Science Foundation of Shandong Province(No.ZR2020ZD35),the Shandong University Multidisciplinary Research and Innovation Team of Young Scholars(No.2020QNQT015),the Young Scholars Program of Shandong University(YSPSDU,No.2018WLJH48),and the Fundamental Research Funds of Shandong University(No.2017TB003).The scientific calculations in this paper have been done on the HPC Cloud Platform of Shandong University.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.02.039.
 Chinese Chemical Letters2022年12期
Chinese Chemical Letters2022年12期
- Chinese Chemical Letters的其它文章
- Diverse strategic approaches en route to Taxol total synthesis
- Recent advances in gold-complex and chiral organocatalyst cooperative catalysis for asymmetric alkyne functionalization
- Unmodified methodologies in target discovery for small molecule drugs:A rising star
- Recent advances in single-crystalline two-dimensional polymers:Synthesis,characterization and challenges
- Environmental applications of graphene oxide composite membranes
- Recent advances in the application of metal organic frameworks using in advanced oxidation progresses for pollutants degradation