
Electrooxidative[3+2]annulation of amidines with alkenes for the synthesis of spiroimidazolines
2023-01-30 06:48SaiZhangGaochenXuHuanYanQinghuanWuJingjingMengJindianDuanKaiGuo
Chinese Chemical Letters 2022年12期
Sai Zhang,Gaochen Xu,Huan Yan,Qinghuan Wu,Jingjing Meng,Jindian Duan,Kai Guo
College of Biotechnology and Pharmaceutical Engineering,State Key Laboratory of Materials-Oriented Chemical Engineering,Nanjing Tech University,Nanjing 211816,China
Keywords:Electrooxidative Amidines Alkenes Spiroimidazolines Annulation
ABSTRACT The electrooxidative[3+2]annulation of amidines with 2-arylideneindane-1,3-diones or 4-alkylidene pyrazolones is reported using NaI as a redox catalyst and electrolyte under constant current electrolysis in an undivided cell.The current strategy features excellent functional group tolerance,simple operation,and mild conditions,thus providing an environmentally benign and efficient access to spiroimidazolines in moderate to good yields.
Spiroimidazolines are basic structural motifs embedded in numerous natural products and pharmaceutical molecules,which have shown a broad spectrum of extraordinary biological properties[1,2],and they are also used as quintessential intermediates for the synthesis and derivatization of more complex molecules[3].However,only a very few reports have been concerned for the synthesis of these spiroheterocyclic motifs[4–6].Therefore,the development of facile and straightforward strategies towards spiroimidazolines is of urgent and highly desirable.
On the other hand,amidines have gained increasing attention as eminently crucial and pivotal synthons in organic synthetic chemistry[7–12].Especially,tremendous efforts have been made for the construction of nitrogen-containing heterocycles through[3+2]annulation of amidines with various alkenes[13–15].In the past decade,Fe or Cu catalyzed coupling reaction of amidines with enals,chalcones,nitroolefins or enaminones have been successfully established to access the structurally diverse imidazoles by Liet al.[16–20],Zhuet al.[21]and Zhanet al.[22](Scheme 1a).More recently,Xuet al.[23–25]reported excellent work on the synthesis of spiroimidazolinesviaNIS or NBS-promoted cyclization of amidines with diverse activated alkenes(Scheme 1b).However,the existing methods suffer from some drawbacks,including the use of transition metal catalysts,stoichiometric amount of corrosive exogenous oxidants and harsh reaction conditions.
Organic electrochemistry represents an environmentally benign and sustainable tool for various organic transformations,avoiding the utilization of external chemical redox reagents due to use of electrons as oxidizing or reducing reagents[26–38],and a series of elegant electroorganic reactions for C–N bond formations have been developed independently by several research groups[39–52].To the best of our knowledge,the synthesis of nitrogencontaining heterocycles from amidinesviaan electrochemical oxidative coupling has never been reported.As a continuation of our research focus on electrochemical transformation and the synthesis of structurally novel spirocyclic compounds[53–55],we present herein our preliminary work on electrooxidative[3+2]annulation of amidines with alkenes for the synthesis of spiroimidazolines under transition-metal catalyst and oxidant free conditions(Scheme 1c).
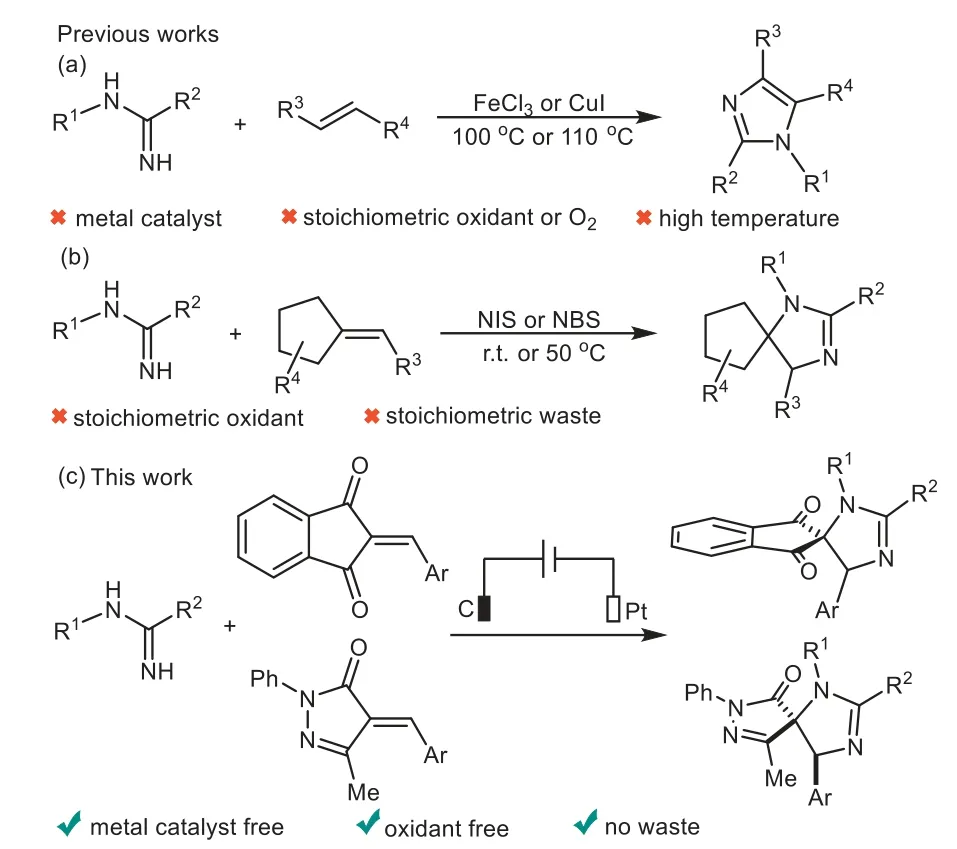
Scheme 1.[3+2]Annulation of amidines with alkenes.
We initially started our investigation by employing 2-benzylidene-1,3-indandione 1a andN-phenylbenzamidine 2a as the model substrates to optimize reaction conditions.Gratifyingly,the desired spiroimidazoline 3aa was obtained in 96%yield by performing the electrolysis at room temperature at a constant current of 5 mA for 5 h in an undivided electrolytic cell equipped with a graphite anode and a platinum plate cathode with a mixed solvent of MeCN/H2O(9/1,v/v)containing electrolyte NaI(Table 1,entry 1).The examination of different supporting electrolytes including NaBr,Me4NI and Bu4NI resulted in inferior outcomes(entries 2–4).Notably,the reaction was completely abolished when Bu4NOAc was chosen as the electrolyte(entry 5).Solvent screening indicated that changing the amount of H2O negatively affected the yields(entries 6–8).A slightly decrease in the chemical yield of 3aa was observed when C or Ni was used instead of Pt as the cathode(entries 9 and 10).The increase or decrease in the electric current afforded the final product in lower yields(entries 11 and 12).Prolonging the reaction time to 10 h did not affect the conversion efficiency(entry 13).Finally,control experiments confirmed the pivotal roles of electric current and electrolyte in enabling this annulation process(entries 14 and 15).
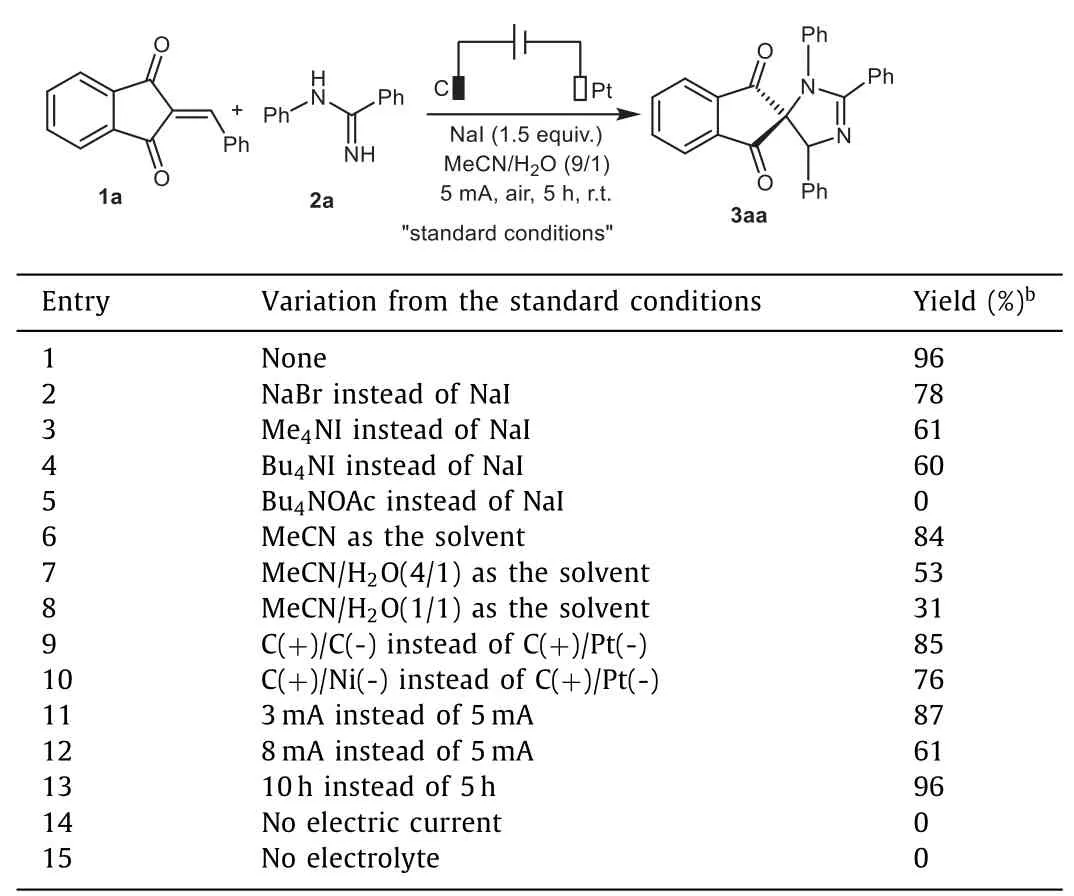
Table 1 Optimization of the reaction conditions.a
With the optimized reaction conditions described above,we then investigated the substrate scope and generality of the electrocatalytic heteroannulation of a variety of 2-arylideneindane-1,3-diones 1 withN-phenylbenzamidine 2a,and the detailed results are summarized in Scheme 2.Gratifyingly,the aromatic rings could be functionalized with an electronically diverse range of substituents at thepara-,meta-,orortho-positions and the substrates with electron-donating substituents(Me,MeO)or electron-withdrawing groups(F,Cl,Br,NO2)were generally well tolerated and transformed to the corresponding products in moderate to high yields(3ba–3la).The results indicated that the electron density and steric hindrance of the phenyl ring did not dramatically influence the reaction.Additionally,the synthetic value of this transformation was further demonstrated with its efficient application to biphenyl,β-naphthyl and thiophenyl-substituted 1,3-indandiones,affording the desired products 3ma–3oa in 83%–93%yields under the standard reaction conditions.However,isopropylsubstituted 1,3-indandione failed to give the desired product 3pa but afforded a messy mixture instead.In addition,chalcone was also not amenable to this procedure.To demonstrate the synthetic utility of this protocol,a scale up experiment was performed,specifically at 5 mmol scale and 3aa was afforded in 82%yield.
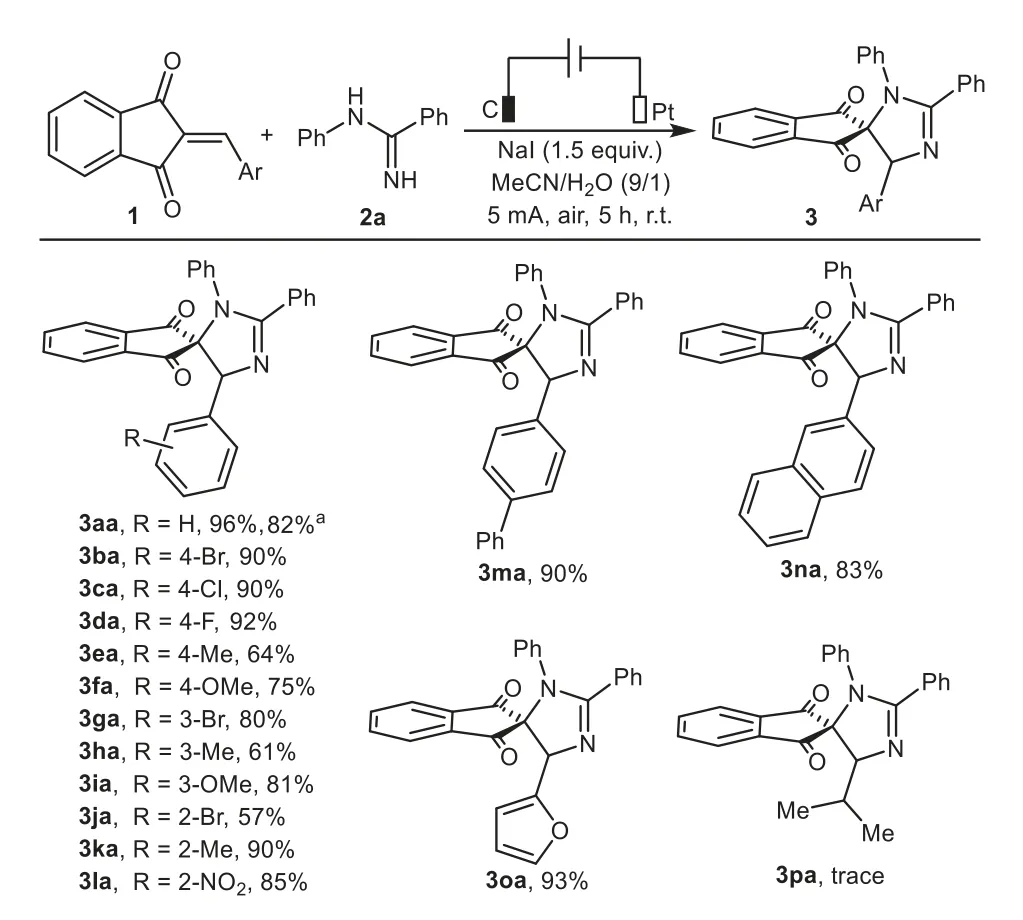
Scheme 2.Substrate scope of 2-arylideneindane-1,3-diones.Reaction conditions:C anode(10 mm×10 mm×3 mm),Pt cathode(10 mm×10 mm×0.2 mm),undivided cell,5 mA,1(0.375 mmol),2a(0.25 mmol),NaI as electrolyte(0.375 mmol),MeCN/H2O(4.5 mL/0.5 mL),air,r.t.,5 h.a 5 mmol scale.
Subsequently,our efforts focused on investigating the scope with respect to differentN-phenylamidines.As the results in Scheme 3 revealed,variousN-phenylbenzamidines,regardless of the electronic properties,steric hindrances,and substitution positions on theN-phenyl ring,could react smoothly with 1a to produce targeted products ranging from 63%to 89%(3ab−3aj).Moreover,substituents such as halogen(F,Cl,Br),methyl,methoxyl at thepara-,meta-,orortho-positions of C-phenyl ring also performed well to generate the corresponding products in 60%–92%yields(3ak−3ap).To our disappointment,the expected products were not successfully afforded when one of the aryl groups was replaced with an aliphatic group(not given in Scheme 3).
This electrocatalytic transformation was also applicable to a wide range of 4-alkylidene pyrazolones bearing electron-donating and electron-withdrawing substituents on the phenyl ring(Scheme 4),which enabled the formation of structurally diverse spiroimidazolines 5a−5j in moderate to good yields(54%−82%)with generally excellent diastereoselectivities(all>20:1).It should be noted that there was no straightforward correlation between the electronic properties of the substituents and reaction efficiency,while increasing steric hindrance posed by theortho-position led to a slightly decrease in reactivity(5h−5j).
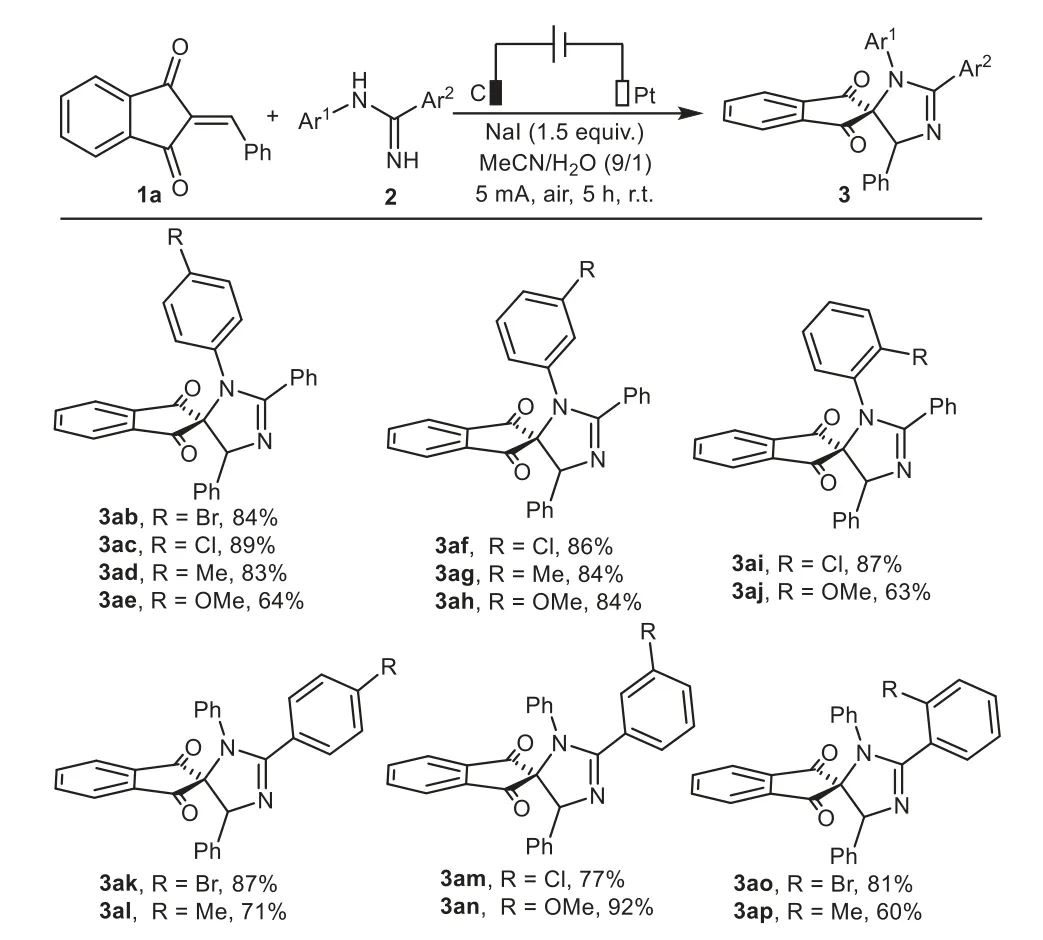
Scheme 3.Substrate scope of N-phenylamidines.Reaction conditions:C anode(10 mm×10 mm×3 mm),Pt cathode(10 mm×10 mm×0.2 mm),undivided cell,5 mA,1a(0.375 mmol),2(0.25 mmol),NaI as electrolyte(0.375 mmol),MeCN/H2O(4.5 mL/0.5 mL),air,r.t.,5 h.
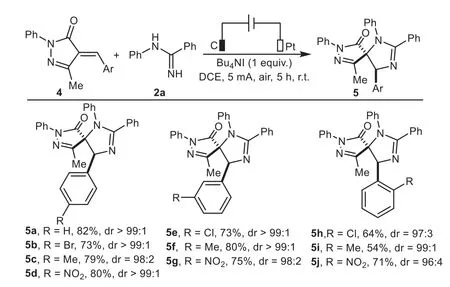
Scheme 4.Substrate scope of 4-alkylidene pyrazolones.Reaction conditions:C anode(10 mm×10 mm×3 mm),Pt cathode(10 mm×10 mm×0.2 mm),undivided cell,5 mA,4(0.5 mmol),2a(0.25 mmol),Bu4NI as electrolyte(0.25 mmol),DCE(5 mL),air,r.t.,5 h.
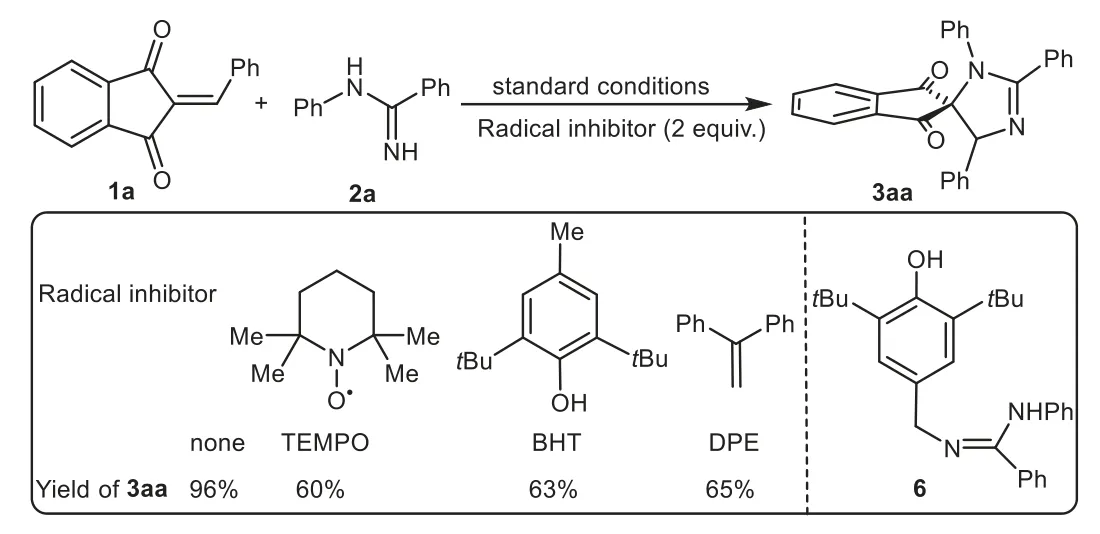
Scheme 5.Control experiments.
To achieve insights into the electrochemical spiroannulation mechanism,several control experiments were carried out(Scheme 5).When the radical scavengers,such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), tertbutylhydroxytoluene(BHT)or 1,1-diphenylethene(DPE)were added to the reaction mixture,the electrochemical process was not completely inhibited but led to a sharp decrease in the yield of 3aa.Furthermore,a BHT-trapped complex 6 was detected by electrospray ionization mass spectrometry(ESI-MS)analysis(see Supporting information for details),suggesting theN-radical intermediate might be involved in this system,which was contrary to the ionic mechanism reported by Xuet al.[23–25].
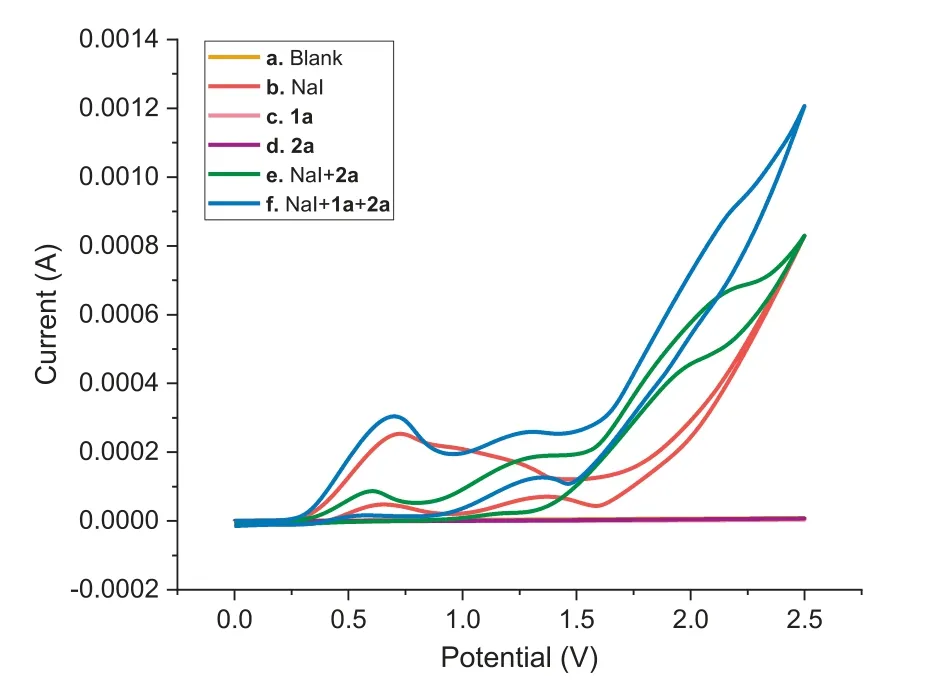
Fig.1.Cyclic voltammograms of reactants and their mixtures in MeCN/H2O(9:1,v/v,10 mL)at room temperature:(a)background;(b)NaI(0.075 mol/L);(c)1a(0.075 mol/L);(d)2a(0.05 mol/L);(e)NaI(0.075 mol/L)+2a(0.05 mol/L);and(f)NaI(0.075 mol/L)+1a(0.075 mol/L)+2a(0.05 mol/L).The voltammogram was obtained at a scan rate of 0.1 V/s with the Pt electrode as a counter electrode,Ag/AgCl(saturated KCl)as a reference electrode,and glassy carbon electrode(diameter,3.0 mm)as a working electrode.
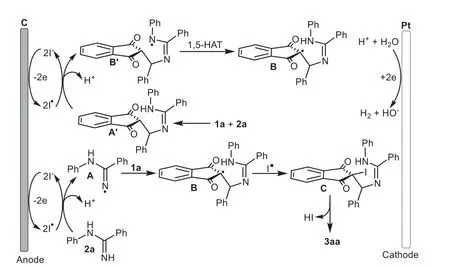
Scheme 6.Plausible mechanism.
We also conducted cyclic voltammograms(CV)experiments to determine the redox behavior of the reactants and their mixtures.As depicted in Fig.1,the CV of NaI(curve b)exhibited an obvious reversible redox wave,with the oxidation peaks at 0.73 VversusAg/AgCl,and substrates 1a and 2a presented no significant oxidation peaks in the range of 0.0−2.5 VversusAg/AgCl(curves c and d)without NaI.Nevertheless,the reduction peak of NaI disappeared along with the generation of a new oxidation peak at 0.60 V in the presence of 2a(curve e).This is the typical behavior of mediator-assisted oxidation,indicating that iodine radicals promoted the production ofN-radicals and the process involved indirect electrolysis[56–61].Moreover,a weak oxidative peak at 0.70 V was observed in a mixture of NaI,1a and 2a(curve f),implying another oxidative process.
Based on the above experimental data and previous reports[57,58],a possible mechanism was outlined and described in Scheme 6.Initially,an iodine anion underwent oxidation to generate the iodide radical at the anode.Next the interaction ofNphenylbenzamidine 2a with the iodide radical formed theN-radical intermediate A,and then the radical addition of 1a generated a carbon radical intermediate B.Alternatively,radical B could also be formed via the oxidation of iodide radical to thein situ-generated aza-Michael adduct A’followed by the intramolecular 1,5-HAT process.Subsequent radical coupling with an iodide radical furnished the halogenated intermediate C,followed by an intramolecular nucleophilic substitution to afford the final product 3aa along with the release of HI.At the same time,cathodic reduction of protons led to the formation of H2.
In conclusion,we have developed the electrooxidative[3+2]annulation of amidines with 2-arylideneindane-1,3-diones or 4-alkylidene pyrazolones for the synthesis of spiroimidazolines in moderate to good yields under transition-metal catalyst and oxidant free conditions.Moreover,the present electrochemical transformation features excellent functional group tolerance,simple operation,and mild conditions.Further studies on the scope and application of the heteroannulation process are currently underway in our laboratory.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We gratefully acknowledg the National Natural Science Foundation of China(No.22078150)and the Natural Science Foundation of Jiangsu Province,Frontier Project(No.BK20212003)for their financial support.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.04.006.
 Chinese Chemical Letters2022年12期
Chinese Chemical Letters2022年12期
- Chinese Chemical Letters的其它文章
- Diverse strategic approaches en route to Taxol total synthesis
- Recent advances in gold-complex and chiral organocatalyst cooperative catalysis for asymmetric alkyne functionalization
- Unmodified methodologies in target discovery for small molecule drugs:A rising star
- Recent advances in single-crystalline two-dimensional polymers:Synthesis,characterization and challenges
- Environmental applications of graphene oxide composite membranes
- Recent advances in the application of metal organic frameworks using in advanced oxidation progresses for pollutants degradation