
Integrating aryl chlorides into nickel-catalyzed 1,1-difunctionalization of alkenes
2023-01-30 06:48CaocaoSunGuoyinYin
Chinese Chemical Letters 2022年12期
Caocao Sun,Guoyin Yin
The Institute for Advanced Studies,Wuhan University,Wuhan 430072,China
Keywords:Aryl chlorides Difunctionalization of alkenes Regioselectivity Nickel catalysis
ABSTRACT Difunctionalization of alkenes have developed into an important type of reactions for rapidly and effi-ciently assemble complex molecules.While extensive advancements have been achieved by the assistance of transition metal catalysis,the employment of cheap,abundant aryl chlorides as coupling partner is still a challenging task in this field.Herein,we report our first achievement in 1,1-difunctionalization of alkenes with aryl chlorides as coupling partners.The success is predominantly ascribed to the judicious selection of 1,2-diamine ligand.This study provides an efficient protocol for the synthesis of secondary benzyl boronates from easily accessible feedstock chemicals.Furthermore,the distinguished features of this method include excellent 1,1-regio-and chemoselectivity,good functional group tolerance and easilyoperational catalytic reaction conditions.
Alkenes are one of most abundant fine chemical products in petroleum industry[1,2].Difunctionalization of alkenes,by opening the double bond of alkenes to form two new chemical bonds simultaneously,represents an efficient means to rapidly construct complex C(sp3)-riched functional molecules[3–9].Electrophilic aryl halides are always employed as the aryl fragment precursors in these transformations[10–15].Although extensive progresses have been made in cross-coupling of aryl chlorides,by the means of either transition metal catalysis[16–28]or transition metal free catalysis[29–35],the state-of-the-arts of alkene difunctionalizations still highly reply on the application of aryl iodides and bromides as the electrophilic aryl coupling partners.The advantages of utilizing aryl chlorides as the aryl precursors mainly include following two facets:(1)Aryl chlorides are relative abundant electrophiles compared with their iodides and bromides counterparts(Scheme 1a);(2)Aryl chlorides are always well-tolerated in many transformations,allowing them to be introduced in early steps and transferred later.The challenge of integrating aryl chlorides into transition-metal catalyzed difunctionalization of alkenes principally derives from the contradiction between their low reactivity toward oxidative addition and the readilyβ-H elimination of formed alkylmetal intermediates(Scheme 1b)[36].As a continuation of our interest in alkene transformations[37–45],particularly after the success in a series of migratory carboboration reactions[37–42],we moved our attention to the challenge of integrating aryl chlorides into alkene difunctionalizations.Herein we describe our successful realization in this goal,by the development of three-component,nickel-catalyzed 1,1-arylboration of unactivatedα-alkenes with aryl chlorides as electrophilic coupling partners(Scheme 1c).
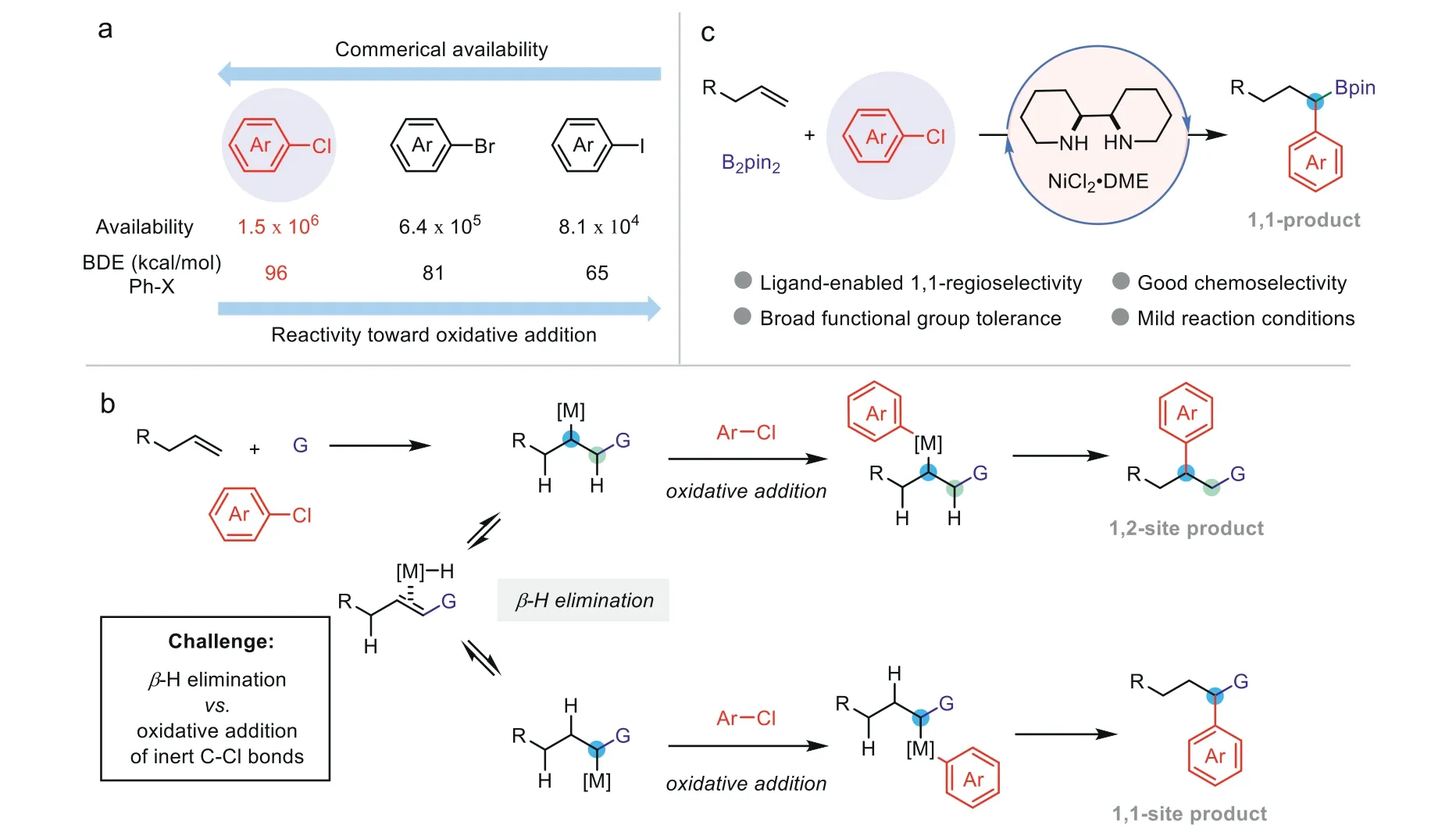
Scheme 1.Difunctionalization of alkenes with aryl chlorides.(a)Comparison of aryl chlorides with their analogues.(b)Challenges of di functionalization of alkenes with aryl chlorides.(c)Nickel-catalyzed 1,1-arylboration of undirected,unactivated alkenes with aryl chlorides(this work).
We commenced this study by choosing 1-octene (1a),bis(pinacolato)diboron(2)and phenyl chloride(3a)as model substrates.Reaction optimization was conducted next,and selected results have been listed in Table 1.The first tentative attempt with the same reaction conditions[37]at an elevated temperature(80°C)gave the 1,1-arylboration product 4a in 15%yield with excellent regioisomeric ratio(rr>20:1)(entry 1).Following solvent survey indicated that polar solvents,such as DMA,DMF and DMSO,were ineffective for this 1,1-difunctionalization reaction(entries 2 and 3).Only small amount of 4a could be detected in dichloroethane(entry 4).It should be noted that the solvent could affect the regioselectivity of reaction[46–49],as poor regioselectivity(rr=1:1)was obtained in isopropanol(entry 5).To our delight,the yield could be improved to 36%when the reaction was conducted in ethyl acetate(entry 6).Evidently,rational selection of ligand is critical for the reactivity,therefore an effort was given to ligand screening next.Modifying the structure of 1,2-diamine was found that only symmetriccis-2,2′-bipiperidine(L7)could effectively promote the formation of 1,1-addition product 4a(entry 8),while itstrans-isomer was ineffective for this transformation(entry 7).Poor yield with only moderate enantiomeric excess value(72%ee)was obtained when(R,R)-L7 was used.However,the employment of a commercially available mixture ofcis/trans-isomers(L8)could deliver a similar reactivity,by affording the product 4a in 72%isolated yield with an excellent regioselectivity(rr>20:1)on 0.5 mmol scale(entry 9).The significance of ligand choice was further enhanced by all other types of bisnitrogen-ligated ligands,including those structures based on pyrox(L11),bioxolines(L12),2,2′-bipyridine(L13)and 1,10-phenanthroline(L14)were all ineffective for this transformation(please see Supporting information for details),with the detection of 1,2-diborylation product 4a’and/or Miyaura borylation product 4a”as main by-products.Base surveying discovered that only LiOMe and LiOH could promote the three-component reaction(entries 9–14).It is particularly notable that,these results suggested that the regioselectivity of this reaction was not controlled by the directing group of the substrate.Control experiments revealed that no more than trace amount of 4a was detected in the reaction either without ligand,nickel salt or base(entries 15 and 16).
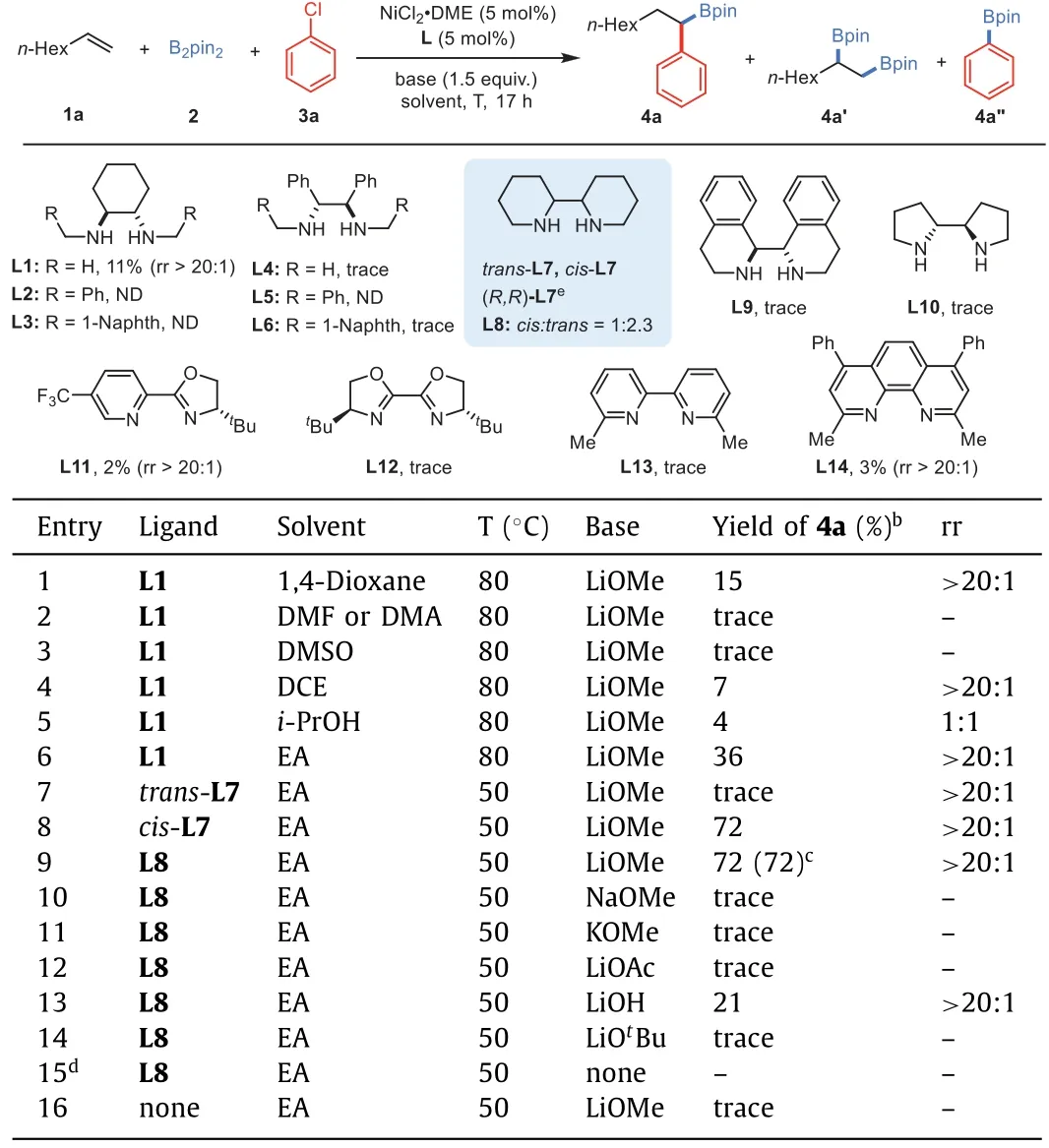
Table 1 Optimization of reaction conditions.a
With the optimal reaction conditions in hand,we next shifted our attention to the generality investigation of this threecomponent reaction.A number of aryl chlorides bearing various substitution patterns and unactivated alkenes containing a wide scope of functional groups were examined,and the results are illustrated in Scheme 2.The reactivity is not largely affected by the electronic property of the aryl electrophilic partners,as the corresponding secondary benzyl boronates could be obtained from both electron-deficient and-rich aryl chlorides,albeit lower yields generally obtained in the cases of the electron-rich ones(4c and 4j).Unactivated alkenes tethering ethers(4t,4v,4w-4y),tosylate(4u),alkylbromide(4n),imide(4ah and 4ai)and esters(4z-4ag)could participate in this arylboration reaction to produce the desired products in good yields with a good regioisomeric ratio(rr>20:1).Moreover,good 1,1-regioselectivity should also be obtained in the reactions with allylsilane(4aj and 4ak)and allylamine derivative(4al).A wide scope of transformable functional group includes amines,carboamides,imide,esters,phosphonate,arylboronate,and ketone were all well compatible in these mild nickel-catalyzed reaction conditions.In addition,the practicability of this reaction was demonstrated by that no significant decrease yield was obtained in a scale-up experiment(4a).
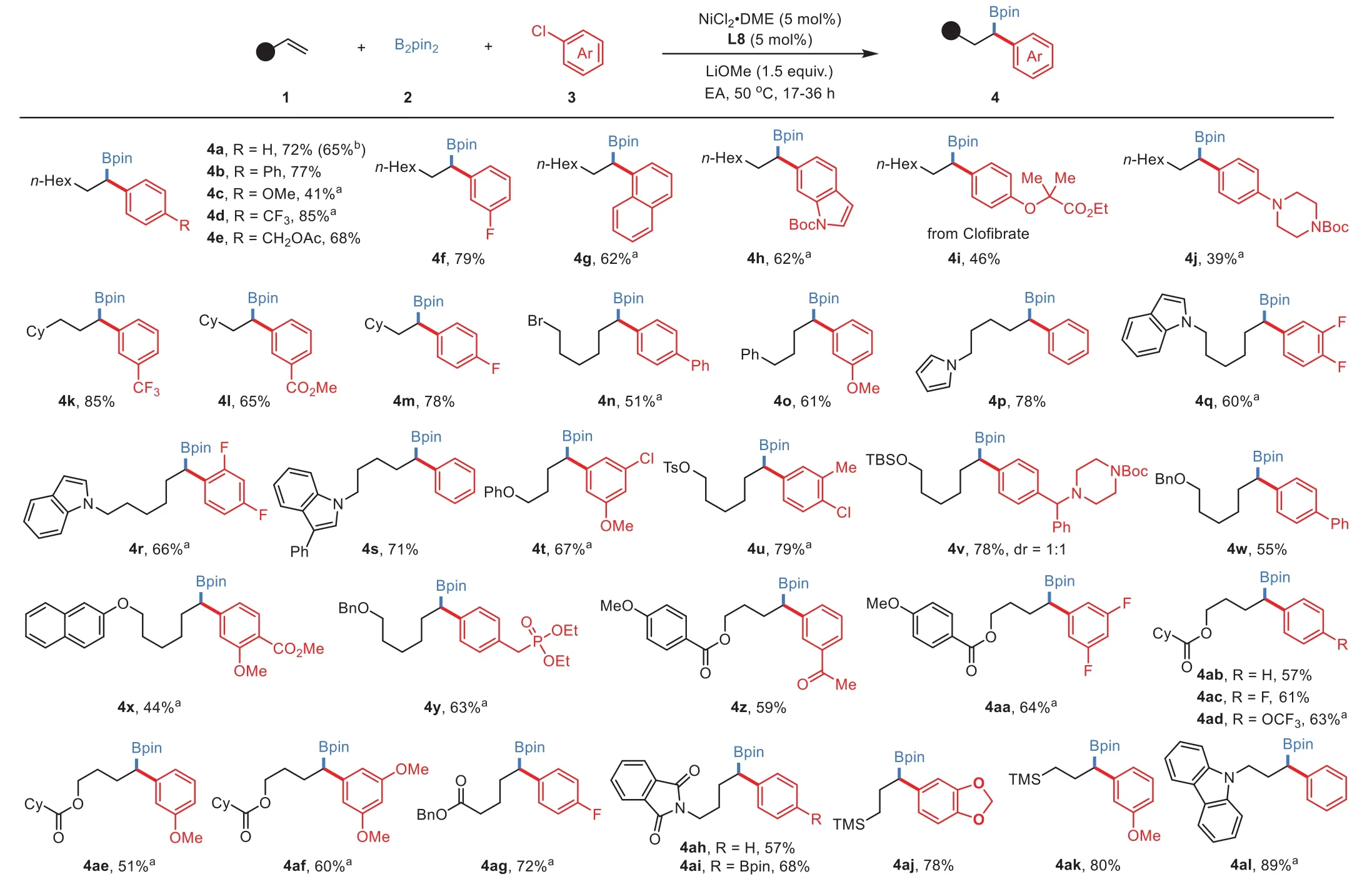
Scheme 2.Study on reaction scope.General reaction conditions:NiCl2·DME(5 mol%),L8(5 mol%),1(0.5 mmol,1.0 equiv.),B2pin2 2(1.5 equiv.),3(1.5 equiv.)and LiOMe(1.5 equiv.)in EA(2 mL),stirred for 17–36 h.Isolated yield.a Isolated yield of the corresponding alcohol after oxidation.b Isolated yield on 5 mmol scale.
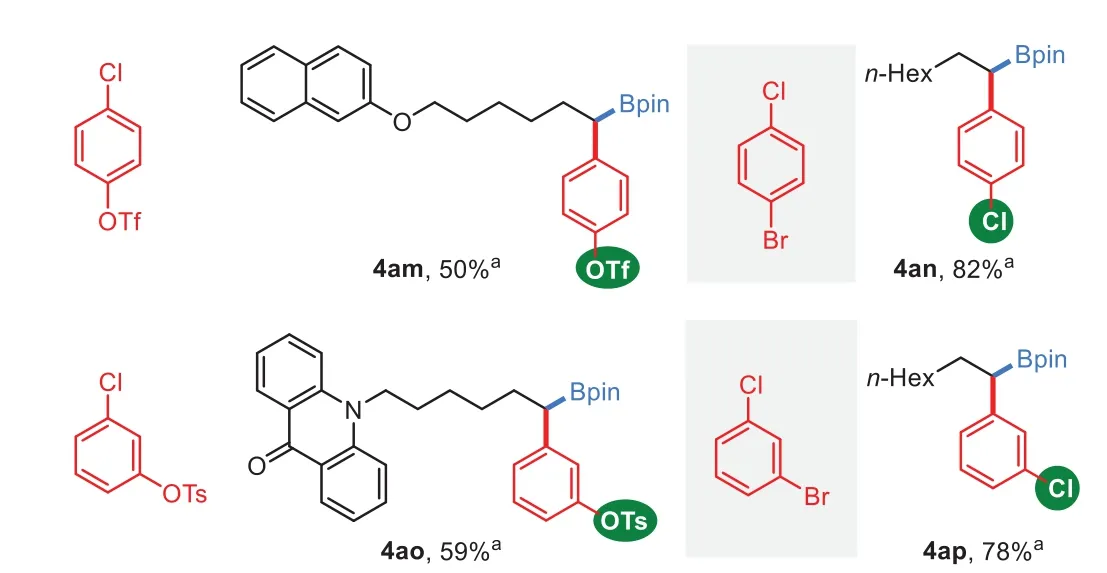
Scheme 3.Chemoselectivity reaction of aryl electrophiles.General reaction conditions:NiCl2·DME(5 mol%),L8(5 mol%),1(0.5 mmol,1.0 equiv.),B2 pin2 2(1.5 equiv.),3(1.5 equiv.)and LiOMe(1.5 equiv.)in EA(2 mL),stirred for 36 h.Isolated yield.a Yield of the corresponding alcohol after oxidation.
To further explore the synthetic potential of this method,chemoselectivity of multi-(pseudo)halogenated arenes were investigated.As shown in Scheme 3,C(sp2)-Cl bond is reactive when either C(sp2)-OTf bond(4am)or C(sp2)-OTs bond(4ao)existed.However,the C(sp2)-Cl bond is inert when it coexists with C(sp2)-Br bond(4an and 4ap).The resting(pseudo)halides open avenues for manipulations by further cross-coupling events[50–57].Moreover,when allylbenzene derivatives were used,both 1,1-and 1,3-site addition products[58–61]could be selectively obtained by only alternation of the ligand(Scheme 4).The less sterically hindered diamine ligand L8 is favored the C–C bond formation atα-position of boron group,while more sterically hindered 1,2-diamine ligand L6 favored the arylation at benzylic position(see Supporting information for results of more ligands).Notably,the regioselectivity of these reactions were slightly affected by the utilized electrophiles.
To shed light on the catalytic cycle of this Ni-catalyzed threecomponent reaction,mechanistic experiments were designed and carefully implemented.First,a terminal deuterium-labeled alkene 5f-dwas synthesized and examined under the standard reaction conditions,which afforded the 1,1-addition product 6f-din 32%(Scheme 5a).Deuterium was observed at the bothα-andβposition,but was not detected at theγ-position.Moreover,when excess amount of methanol was added to the reaction with 5f and 3d,the formation of product 6f was inhibited and hydroboration byproduct 8 was increased(Scheme 5b,comparison of entries 1 and 2).The product 8 could be isolated,but without deuterium incorporation into the benzylic position,when CD3OD was employed(Scheme 5b,entries 3 and 4).These findings suggest thatβ-hydride elimination is selective at theα-position of boronic ester group,even in presence of a competing benzylic group,under current catalytic system.
Finally,we proposed a catalytic cycle for this nickel-catalyzed 1,1-arylboration reactions based on the above discoveries and related precedent studies[62–64].As outline in Scheme 5c,a L8-ligated nickel chloride catalyst(I)is formed and initialed the reaction.In the presence of LiOMe,transmetallation leads to the formation of a nickel methoxylate catalyst II,and further transmetallation with B2pin2affords a Ni-Bpin species III.Then olefin-ligated intermediate IV was formed after olefin coordination.Following migratory insertion selectively in ananti-Markovnikov manner,leads to the formation of intermediate V,rather than V’.Selectiveβ-hydride elimination at theα-position of boronic ester group and following migratory insertion generates a boron group stabilized alkyl nickel intermediate VI.The intermediate VI reacts with the aryl chloride to deliver the 1,1-arylboration product 4 and regenerate the catalyst I.
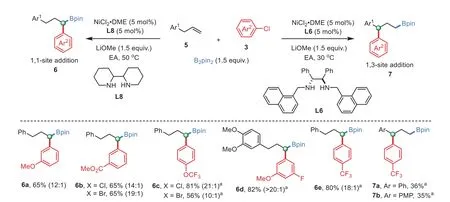
Scheme 4.Regioselectivity reaction of allylbenzenes.General reaction conditions:NiCl2·DME(5 mol%),L(5 mol%),5(0.5 mmol,1.0 equiv.),B2 pin2 2(1.5 equiv.),3(1.5 equiv.)and LiOMe(1.5 equiv.)in EA(2 mL),stirred for 17–36 h.Isolated yield.a Yield of the corresponding alcohol after oxidation.
In summary,we have developed the first aryl chlorides participated 1,1-functionalzation of undirected,unactivated alkenes.The application of 1,2-diamine-ligated nickel catalyst governs both the reactivity and selectivity.This study provides a protocol for the efficient synthesis of secondary benzylic boronic esters from cheap,abundant starting materials.We believe this chemistry will greatly benefit to the synthesis of alkyl boron compounds and promote the development of synthetic methodology based on chain-walking.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China(No.22122107)and the Fundamental Research Funds for Central Universities(No.2042021kf0190).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.04.026.
 Chinese Chemical Letters2022年12期
Chinese Chemical Letters2022年12期
- Chinese Chemical Letters的其它文章
- Diverse strategic approaches en route to Taxol total synthesis
- Recent advances in gold-complex and chiral organocatalyst cooperative catalysis for asymmetric alkyne functionalization
- Unmodified methodologies in target discovery for small molecule drugs:A rising star
- Recent advances in single-crystalline two-dimensional polymers:Synthesis,characterization and challenges
- Environmental applications of graphene oxide composite membranes
- Recent advances in the application of metal organic frameworks using in advanced oxidation progresses for pollutants degradation