
Rhodium catalyzed asymmetric synthesis of Chiraphos derivatives
2023-01-30 06:48YingYingSunBinZhngLinginYuRnrnCuiQingyngZhoQingWeiZhng
Chinese Chemical Letters 2022年12期
Ying-Ying Sun,Bin Zhng ,Lingin Yu,Rnrn Cui ,Qingyng Zho ,*,Qing-Wei Zhng ,*
a Department of Chemistry,University of Science and Technology of China,Heifei 230026,China
b School of Pharmac eutical Sciences(Shenzhen),Shenzhen Campus of Sun Yat-sen University,Shenzhen 518107,China
Keywords:Rhodium Phosphine Diene ligand 1,4-Addition Chiraphos
ABSTRACT Herein,we report a highly efficient versatile synthetic route to Chiraphos derivatives through Rh/Ph-bod catalyzed asymmetric addition of aryl boronic acids to phosphinyl dienes.Various substituted phosphinyl dienes,both on the parent skeleton and the phosphine atoms,were well tolerated with this method and provided chiral phosphine oxides in satisfied yield and up to 95%ee.The corresponding Chiraphos derivative displayed an advantage over Chiraphos in the representative Pd-catalyzed asymmetric 1,4-addition reaction.
Biphosphines ligands have found wide application in asymmetric catalysis as ligands in transition metals and organocatalysts by themselves[1–13].Among them,Chiraphos ligand has a pivotal position in catalytic asymmetric hydrogenation[14–17],1,4-addition[18–23]and allyl alkylation reaction[24,25].What is more,it has also been employed by BASF company as a ligand for the industrial asymmetric synthesis of L-menthol[26],which is widely used in pharmaceutical and food science.It has also been used as a chiral ligand for functional transition metal complexes and as an auxiliary for the synthesis of Au24clusters with inherent chirality[27].
However,up to date,the synthesis of Chiraphos mainly relies on the original strategy developed by Bosnichet al.through nucleophilic substitution reactions of chiral 2,3-butandiol derivatives and lithium diphenylphosphide[28–31].On the other hand,Phosphinyl diene 1a has also been used as a more accessible alternative starting material to access Chiraphos ligand.In 1997,Matteoliet al.reported a chiral resolution method through sequential NaBH4-mediated reduction of 1a,resolution of the resulting racemic mixtures and reduction of P-O(Scheme 1a)[32].In 1999,Matteoli’s group achieved a catalytic asymmetric synthesis of ChiraphosviaRu-BINAP catalyzed hydrogenation of 1a.However,the reaction proceeds with harsh reaction conditions(310 h,100°C,100 atm H2)and afforded Chiraphos in low yield and unsatisfactory selectivity(71%ee,70%de)(Scheme 1b)[33].In addition,there are no cases of backbone modify for preparing an array of Chiraphos derivatives in above-mentioned reports,which indeed limits the structure−activity relationship study of Chiraphos and its application in asymmetric catalysis.
On the basis of previous work[34–36],we reported herein a Rhodium catalyzed Hayashi-Miyaura reaction for the efficient synthesis of Chiraphos derivatives(Scheme 1c)[37–42].Phosphinyl diene 1a andp-bromophenyl boronic acid 2a were selected as the model substrate(Table 1).Originally,the reaction was carried out with a rhodium biphosphine catalyst in the presence of a base.However,the reaction was sluggish even at 100°C,while the racemic reaction without a biphosphine ligand proceed smoothly at room temperature.Consequently,a series of diene ligands L1-L6 were tested as an alternative[43–57].To our delight,all of these ligands could give the desired product in excellent yields and highee(entries 1-6,86%-99%yield,62%-84%ee).Among them,(R,R)-Phbod ligand L6 gives the highest enantioselectivity and yield(entry 6,84%eeand 99%yield).When the weaker base K2CO3was used instead of NaOH,a slight decrease in enantioselectivity and yield was observed(entry 7,82%eeand 97%yield).Besides,the screening of solvents indicated that the polarity of the solvent played a key role in determining the enantioselectivity of the reaction(entries 8-11).Generally,the reaction accelerated in polar solvents probably because of better solubility of 1a while provided loweree.Finally,the enantioselectivity could be further improved at 15°C and gave the product in 80%isolated yield and 87%ee(entry 12).
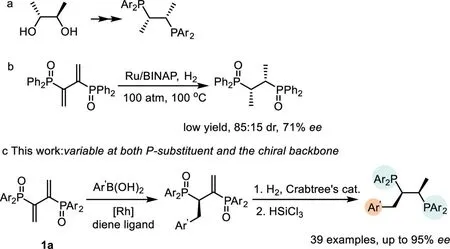
Scheme 1.Methods for the synthesis of Chiraphos derivatives.
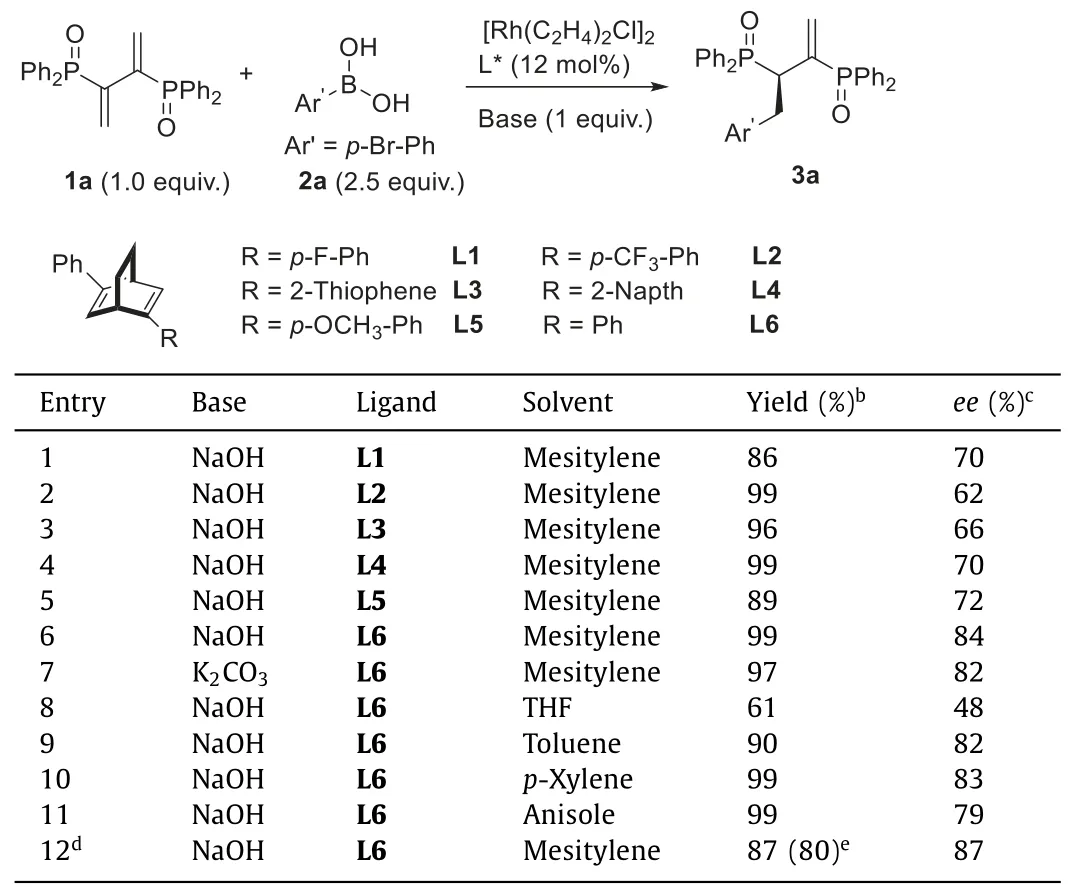
Table 1 Optimization of reaction parameters.a
With the optimized catalytic conditions,we evaluated the scope of the phosphinyl diene substrates(Scheme 2).The stereohindrance of the aryl substituents of the diene substrates could significantly influence the enantioselectivities of the reaction(3a-3e).Atert-butyl group at thepara-position would lead to a dramatic decrease in theee(3d,15%ee).While the electronic properties have little effect both on the enantioselectivity and reactivity,all of the substrates except 3d could deliver the desired products in comparable yields andee(64%-89%).Among these examples,3,5-dimethyl substituted aryl phosphinyl diene could give the product 3h in highest 89%ee.It is worth noting that substrates with a 2-naphthyl or piperonyl substituents could also afford products 3i and 3j in respective 59%,56%yields and 78%,74%ee,thus enabling the synthesis of diversified Chiralphos derivatives.In addition,the bromine atom in the products holds the potential to transform to a series of functional groups by cross coupling reactions.
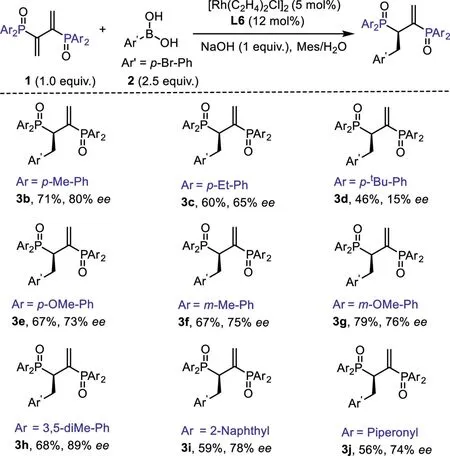
Scheme 2.Substrate scope of phosphinyl dienes.Reaction conditions:0.1 mmol 1,0.25 mmol 2,1 equiv.base,5 mol%[Rh(C2H4)2Cl]2 and 12 mol%ligand in mesitylene/H2O(2 mL,10:1)at 15°C.
The reaction scope was then demonstrated with diphenylphosphinyl(4a-4q)as well as 3,5-dimethyl diphenylphosphinyl(4a’-4r’)dienes in the presence of various arylboronic acids(Scheme 3).
A plethora of substituents of the arylboronic acids atparapositon were tested.In most cases,products from 4a’-4r’could be obtained with over 80%eewhile suffered from lower yields due to inferior reactivities.On the contrary,4a-4q were obtained in high yields albeit with loweree.Electron withdrawing substituents of the arylboronic acids at theparaposition could react smoothly to afford the corresponding products,including halides(4a-4b,4a’-4b’),ester(4c,4c’),aldehyde(4d,4d’),ketone(4e,4e’),trifluoromethyl(4f,4f’)and nitroso groups(4g’).Phenylboronic acid could also deliver the desired products with both phosphinyl dienes in 81%yield(4h)and 92%ee(4h’)respectively.Substrates bearing electron neutral and donatingpara-methyl,para-tertbutyl andpara-methoxyl groups reacted slower,affording the desired products(4i-4k,4i’-4k’)in relatively loweree(65%-82%).The phenomena that electron-withdrawing arylboronic acid reacts more quickly than electron-rich arylboronic acid,indicated that transmetallation was probably the rate-limiting step.Besides,the absolute configuration of 4h has been unambiguously determined to beRby single-crystal XRD analysis(4h,see Supporting information for details).
Substituents at themetaposition of the arlylboronic acids were also tested.All substrates could give the desired products in 67%-85%ee.In addition,2-naphthyl boronic acid could also give the product in 73%ee,albeit with low yield probably due to the stereohindrance of the naphthyl group.The subtle difference in stereohindrance of the boronic acids has a significant influence both on the reactivity and enantioselectivity of the reaction.During the research,we also found that the phosphinyl dienes also suffered from low reactivity in the reaction,as also seen in prof.Matteoli’s paper that harsh reaction conditions were required for the hydrogenation reaction[32].
To investigate the scalability of this reaction,the reaction was carried out at in gram-scale at an elevated temperature(25°C)for higher conversion.As a result,the product 4h was obtained in 85%yield and slightly decreased 76%ee(Scheme 4a).However,theeeof 4h could be easily enriched to 97%by recrystallization from EA in 38%yield.
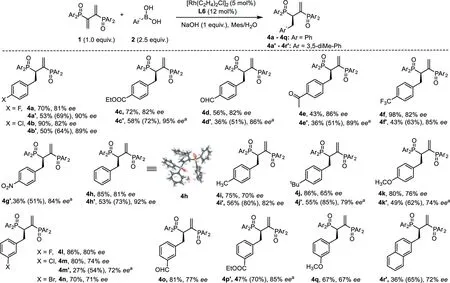
Scheme 3.Substrate scope of arylboronic acids.Reaction conditions:0.1 mmol 1,0.25 mmol 2,1 equiv.base,5 mol%[Rh(C2H4)2Cl]2 and 12 mol%ligand in mesitylene/H2 O(2 mL,10:1)at 15°C or 35°C.Isolated yields based on recovered phosphoryl dienes were shown in parentheses.a Reaction performed at 35°C.
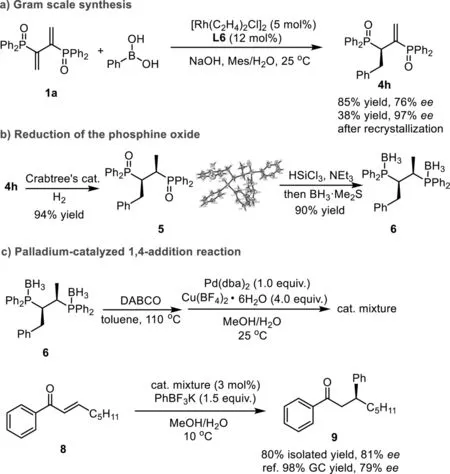
Scheme 4.Gram scale reaction and synthetic application of products.
Furthermore,4h was used as a showcase for facile synthesis of Chiraphos derivative.After extensive study of various reduction conditions,a highly efficient route was developed.As shown in Scheme 4b,the first reduction was performed at the alkene double bond with Crabtree’s catalyst specifically providing 5[58–60]in 94%yield without erosion ofee(97%).The absolute configuration of 5 was unambiguously assigned by single-crystal XRD analysis(5,see Supporting information for details).Subsequently,the second reduction was performed at the P=O with HSiCl3in the presence of triethylamine[29,61].The stable 6 was obtained in one pot with excellent overall yield(90%,two steps),which could give the free disphosphine ligand 7 by removing borane moieties through treating with excess of DABCO[62].
More importantly,in view of the significant role of transition metal catalysis,we successfully evaluated the newly synthesized ligand application in palladium-catalyzed 1,4-addition reaction,this new ligand could give the product 9 with 80%isolated yield and 81%ee(Scheme 4c),which conforming that this kind of modified ligand showed higher enantioselectivity than the parent Chiraphos in the 1,4-addition reaction[31].
In conclusion,a Rh/Ph-bod catalyzed asymmetric addition of aryl boronic acids to phosphinyl dienes has been developed with high yields and good enantioselectivities.The resulting product can be converted to Chiraphos-derivate through a facile route.Moreover,this kind of modified Chiraphos has achieved highly efficient palladium-catalyzed 1,4-addition reactions.Therefore,this strategy enables fine modulating of Chiraphos feasible,and paves a new Chiraphos-based toolbox for transition metal catalyzed asymmetric synthesis.Further investigations toward this field are in progress in our group.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China(Nos.21901235 and 22071224),the USTC Research Funds of the Double First-Class Initiative(No.YD20600002010),and the Anhui Provincial Natural Science-Foundation(No.1908085MB34).Q.Zhao thanks the Science,Technology and Innovation Commission of Shenzhen(No.JCYJ20190807155201669)for the support.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.04.054.
 Chinese Chemical Letters2022年12期
Chinese Chemical Letters2022年12期
- Chinese Chemical Letters的其它文章
- Diverse strategic approaches en route to Taxol total synthesis
- Recent advances in gold-complex and chiral organocatalyst cooperative catalysis for asymmetric alkyne functionalization
- Unmodified methodologies in target discovery for small molecule drugs:A rising star
- Recent advances in single-crystalline two-dimensional polymers:Synthesis,characterization and challenges
- Environmental applications of graphene oxide composite membranes
- Recent advances in the application of metal organic frameworks using in advanced oxidation progresses for pollutants degradation