
Diastereodivergent[4+2]annulation of biphenylenes with enones via nickel(0)-catalyzed C–C bond activation
2023-01-30 06:48JunynChenDchngBiXiuliGuoYiyoWngXingweiLi
Chinese Chemical Letters 2022年12期
Junyn Chen,Dchng Bi ,b,*,Xiuli Guo ,Yiyo Wng ,Xingwei Li ,c,*
a NMPA Key Laboratory for Research and Evaluation of Innovative Drug,Collaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals,Key Laboratory of Green Chemical Media and Reactions,Ministry of Education,School of Chemistry and Chemical Engineering,Henan Normal University,Xinxiang 453007,China
b State Key Laboratory of Organometallic Chemistry,Shanghai Institute of Organic Chemistry,Chinese Academy of Sciences,Shanghai 200032,China
c School of Chemistry and Chemical Engineering,Shaanxi Normal University(SNNU),Xi’an 710062,China
Keywords:Nickel Enones Biphenylenes Diastereodivergent annulation C-C activation 9,10-Dihydrophenanthrenes
ABSTRACT Ni(0)-catalyzed regio-and diastereodivergent[4+2]annulation of biphenylenes withα,βunsaturated ketones is described.This solvent-controlled diastereodivergent reaction integrates C–C bond cleavage of biphenylene and C=C double bond insertion selectivity,offering a mild approach to all possible diastereoisomers of 9,10-dihydrophenanthrene derivatives from the same starting materials.
9,10-Dihydrophenanthrene derivatives are not only ubiquitous motifs in the natural products and bioactive compounds,but also important synthetic intermediates[1–6].For example,Juncusol was isolated from the nature plant and exhibit anticancer and antimicrobial activity[7–9].Cedrelin A was isolated from the bark ofCedrelinga catenaeformisDuke and has cytotoxic activity againstStaphylococcus aureusandBacillus subtilis[10–13].The natural product FD-594 aglycon containing thetransdihydrophenanthrene-diol units(Scheme 1a)[14–16].Although 9,10-dihydrophenanthrene could be synthesized through the transformation of aryns[17–20],transition-metal catalyst annulation reactions[21–26]and others[27–31],the divergent synthesis of these structures from the same starting materials just with different reaction conditions will be much more interesting and attractive.
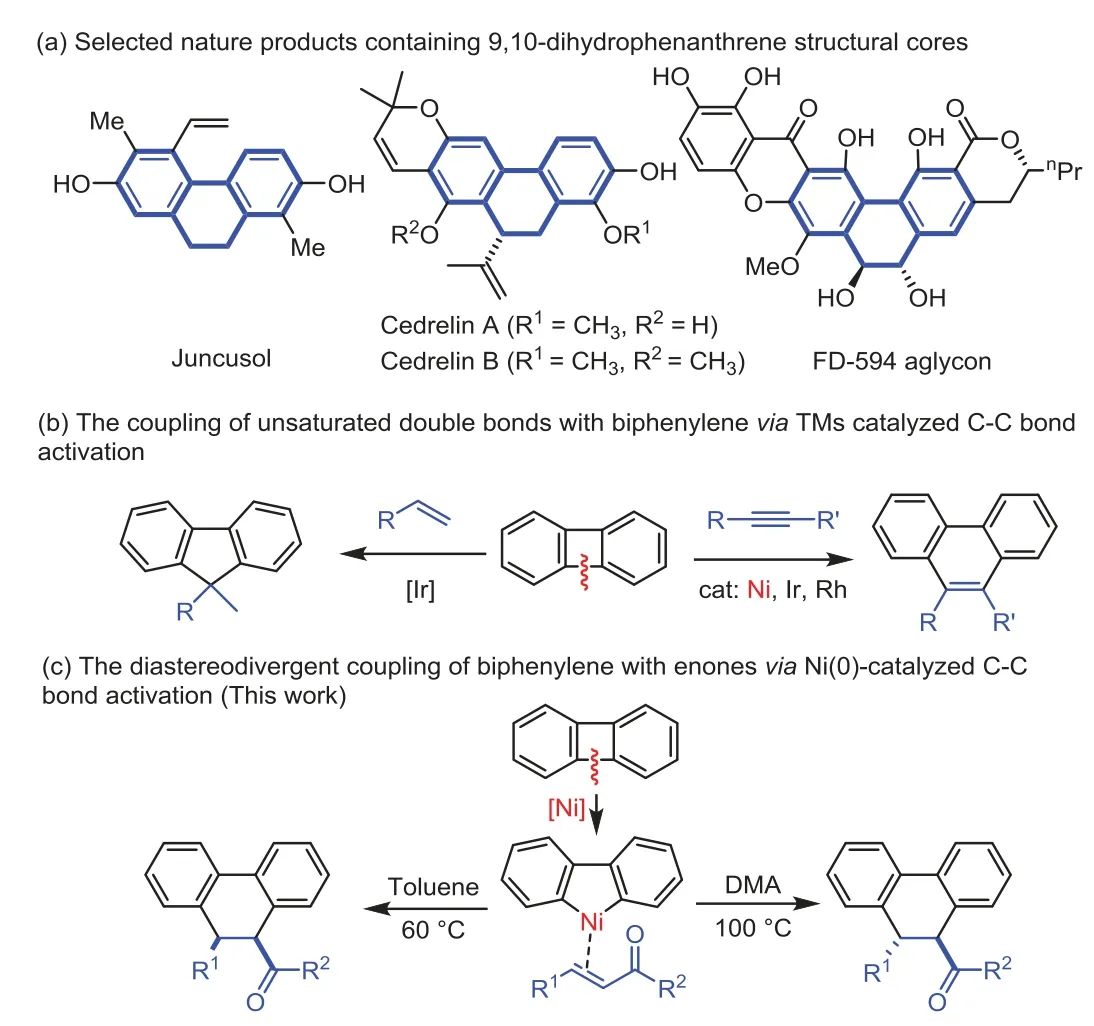
Scheme 1.Transition-metal catalyzed C–C functionalization of biphenylene.
Transition-metal catalyzed C–C bond activation provide an efficient and straightforward approaches for the construction of complex organic structures with perfect atom-and step-economy[32–41].Small strained rings were usually used for the cleavage of C–C bond in presence of transition-metal catalysis,and various attractive synthetic transformations have been reported[36].Biphenylenes are an attractive four-member carbon ring for the C–C bond cleavage and usually used as C4 synthon for the annulation reaction with many types of unsaturated units[42–53].For instance,the alkynes were frequently used in the[4+2]annulation reaction with biphenylene to give phenanthrenes in presence of nickel[42–47],iridium[48]or rhodium catalysis[49].Although,other unsaturated components such as alkenes[50],CO[51]and isocyanides[52,53]were also successfully applied in the ring-opening of biphenylene,the intermolecular coupling of unsaturated double bonds to give the 9,10-dihydrophenanthrene derivatives is still undeveloped(Scheme 1b).In 2016,the Shibata group developed the iridium-catalyzed C–C activation of biphenylenes and coupling with alkenes to give[4+1]cycloaddition products[54].As part of our continuing interest in the intermolecular coupling system through C–C activation using low-cost earth-abundant nickel catalysis[55–57].Herein,we report the nickel-catalyzed diastereodivergent formal[4+2]annulation between biphenylenes and enones,where the C=C bond participated regioselectively to affordtransorcisdiastereoisomer of 9,10-dihydrophenanthrenes just with different reaction conditions(Scheme 1c).Despite the tremendous progress developed in the transition-metal catalyst C–C bond activation,such solvent-controlled diastereodivergent coupling systems for the diverse synthesis of small organic molecular is still rarely reported.This diastereodivergent system would increase the competitiveness and sustainability of transition-metal catalyzed C–C activation.
General procedure A:The mixture of 1a(0.10 mmol),2(0.10 mmol), Ni(cod)2(1.4 mg, 0.005 mmol), PPh3(2.6 mg,0.01 mmol),and DMA (1.0 mL)were charged into a reaction tube.The reaction mixture was stirred at 100°C heated by metal sand bath for 18 h.After the reaction completed(18 h),the reaction mixture was extracted with ethyl acetate(2.0 mL)and saturated NaCl aqueous solution(3×4.0 mL).The combined organic layers were dried over Na2SO4and filtrated,concentrated,and purified by silica gel chromatography using PE/EA(10:1)to affordtrans-3.
General procedure B:The mixture of 1a (0.10 mmol),2(0.10 mmol), Ni(cod)2(1.4 mg, 0.005 mmol), PPh3(2.6 mg,0.01 mmol),and toluene(1.0 mL)were charged into a reaction tube.The reaction mixture was stirred at 60°C heated by metal sand bath for 18 h.After the reaction completed(18 h),the reaction mixture was filtered through a pad of celite,eluted with ethyl acetate,concentrated,and purified by silica gel chromatography using PE/EA(10:1)to affordcis-3.
We selected biphenylene 1a and chalcone 2aa as model substrates to explore the reaction parameters(Table 1).The issues of both chemo-and diastereoselectivity arised when coupling with unsaturated alkenes comparing to the alkynes.It was found that the reaction delivered to the double bond insertion selectivitytrans-product 3aa in 90%yield and excellent diastereoselective ratio(trans/cis>20:1)with Ni(cod)2/PPh3catalysis using the DMA as solvent(entry 1).When decreasing the temperature to 60°C in the DMA solvent,thetrans/cisvalue decreased to 9:1(entry 2).Further study showed that the solvent effect was dramatic(entries 3–9).Low diastereoselective ratio was obtained when the DMF was used as solvent(entry 3).The starting materials were recovered with DCE as solvent(entry 4).Very interestingly,the DME,MeOtBu,THF or dioxane gave another isomer of the[4+2]annulation productcis-product 3aa in excellent yield,even with low diastereoselective ratio(entries 5–8).To our delight,excellent diastereoselectivity could be obtained forcis-3aa when the toluene was used as solvent(entry 9,72%yield,trans/cis=1:9),higher yield anddrvalue were obtained when the reaction temperature decreased to 60°C(entry 10,88%yield,trans:cis=1:>10),further decrease the temperature to 30°C in toluene led to low efficiency(entry 11).Up to now,the solvent-controlled diastereodivergent[4+2]cycloaddition system was established,in which thetrans-3aa was obtainedin polar solvent DMA(entry 1,90%yield,trans:cis>20:1)andcis-3aa was obtained in toluene(entry 10,88%yield,trans:cis=1:>10).Noteworthy,these reaction conditions only afforded the C=C insertion products.Other ligands such as Bpy orrac-BINAP did not promote the annulation(entries 12 and 13),and no corresponding products observed without the addition of PPh3(entries 14 and 15).Our control experiments also showed that the Ni(cod)2was essential for the transformation(entry 16).
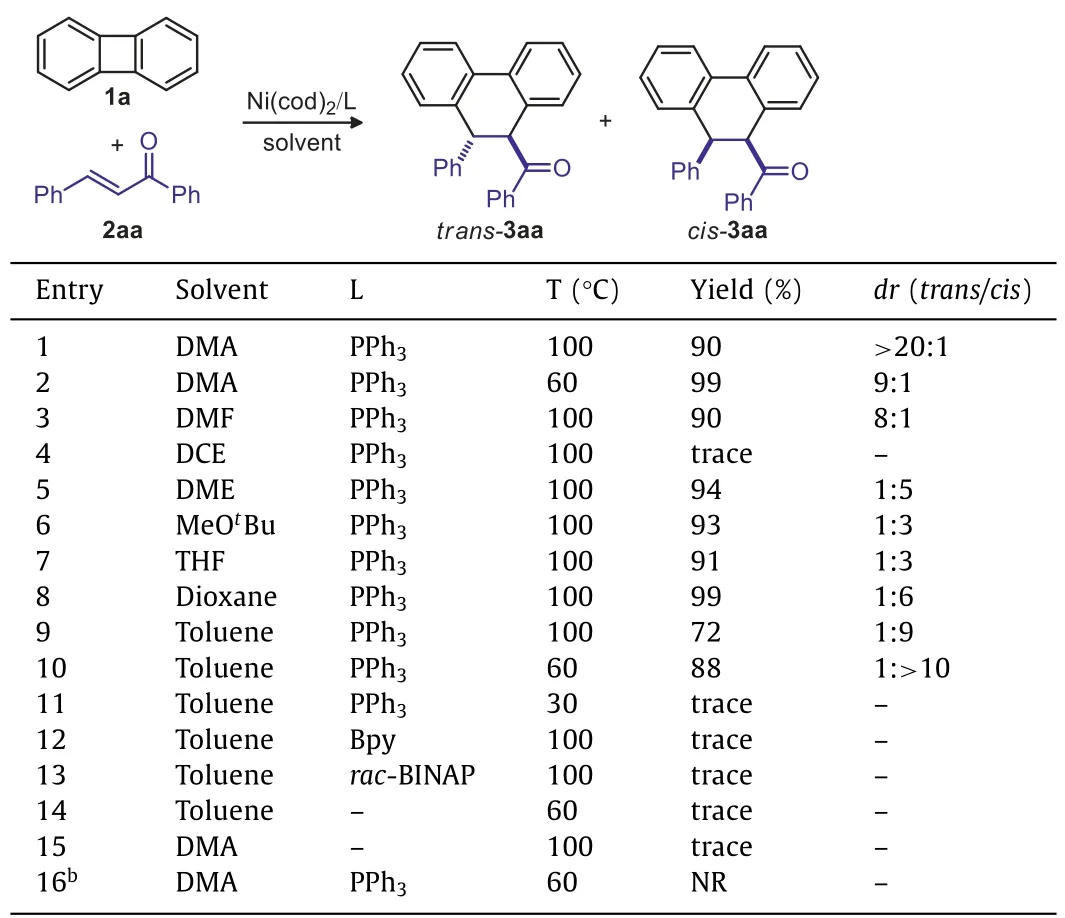
Table 1 Optimization of reaction conditions.a
We next explored the scope of this divergent coupling system(Scheme 2).The generality oftrans-products synthesis was investigated firstly.The reaction tolerated with the electron-donating or-withdrawing groups at theparaposition of the benzene ring in the enones with excellent regio-and diastereoselectivity(trans-3aa totrans-3ae,79%−99%yield,dr=19:1 to>20:1).The configuration oftrans-3ad was determined by X-ray crystallography(CCDC:2116932).The reaction also worked smoothly fororthosubstituted methyl group(trans-3ba,42%yield,dr>20:1)and for pentafluorobenzyl-substituted enones(trans-3ca,58%yield,dr>20:1).The thiophenyl-substituted enone also worked well to give the producttrans-3da in 72%yield and 17:1dr.The reaction for the electron-withdrawing group CF3atpara-position of the benzene ring gave the corresponding producttrans-3ea in 83%yield and>20:1dr.The bromide-substituted enone did not give desired product(trans-3fa,<5%conc.).To our delight,when extension of the chalcones to theβ-CF3substituted enones,the reaction also worked smoothly to give the desired products.Various electrondonating and-withdrawing groups at the different position of the benzene ring tolerated(trans-3ga totrans-3na,41%−99%yield anddr>20:1),When thecis-enone was used,similar results withtrans-enone obtained probably owing to the isomerization ofcis-2ga totrans-2ga in the catalyst systems(trans-3ga,86%yield,dr>20:1).The naphthyl-or furyl-substituted enones all coupled with biphenylene gave the[4+2]annulation product with excellent diastereoselectivity(trans-3oa,84%yield,dr>20:1 andtrans-3pa,71%yield anddr>20:1).Theβ-C2F5substituted enones gave the corresponding products with a slight low yield(trans-3qa,45%yield,dr>20:1 andtrans-3ra,41%yield,dr>20:1).However,the ethyl cinnamate was found to be unreactive.
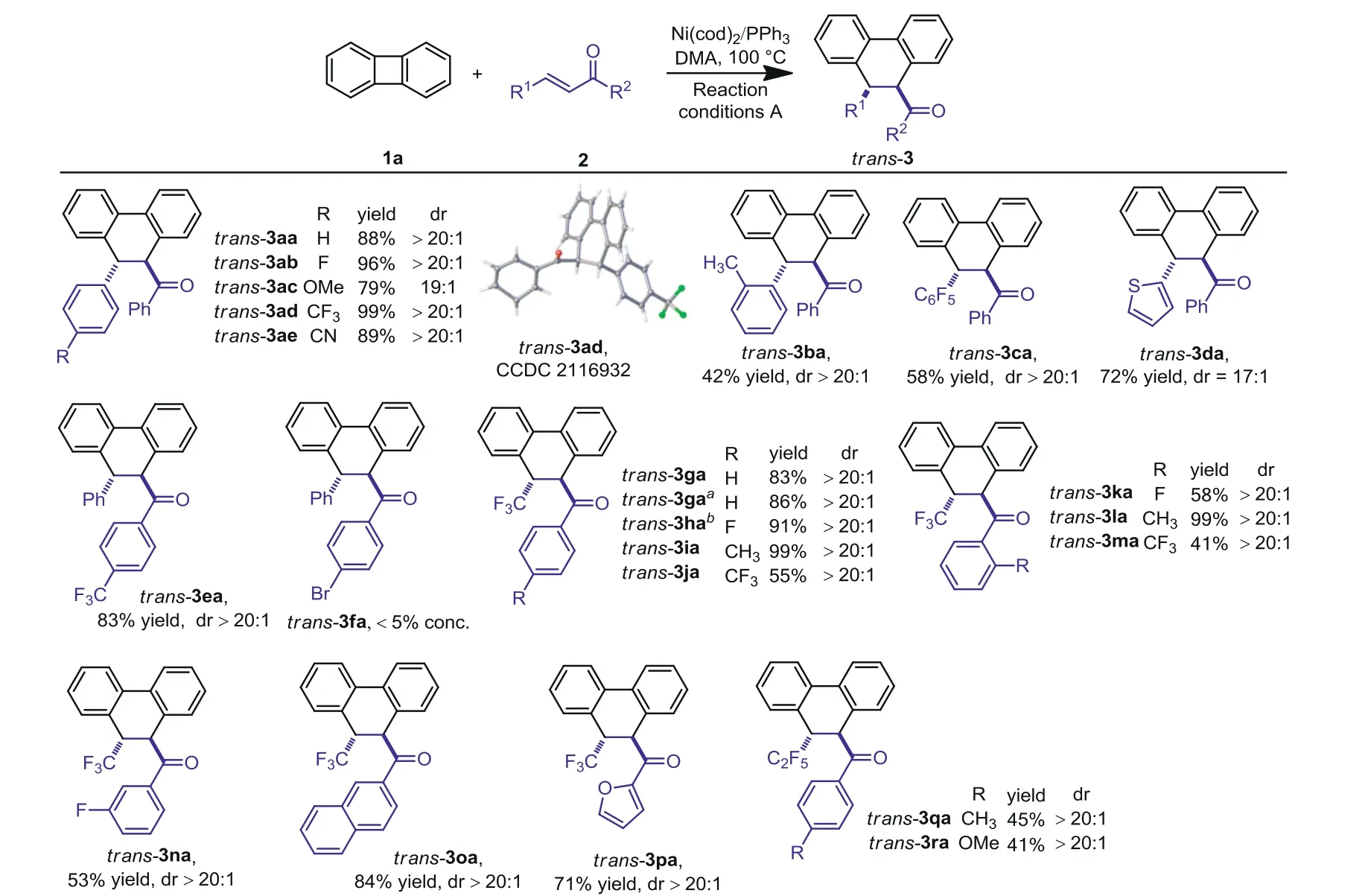
Scheme 2.Scope of substrates for the synthesis of trans-products.Reaction conditions A:Reaction conditions:1a(0.1 mmol),2(0.1 mmol),Ni(cod)2(5 mol%),PPh3(10 mol%),in DMA(1.0 mL),18 h,100°C,argon.a from the cis-enone substrates.b Ni(cod)2(5 mol%),PPh3(5 mol%)in DMA(1.0 mL),dr=the ratio of trans/cis.
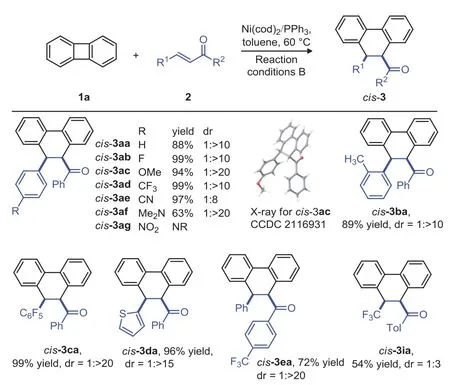
Scheme 3.Scope of substrates for the synthesis of cis-products.Reaction conditions B:1a(0.1 mmol),2(0.1 mmol),Ni(cod)2(5 mol%),PPh3(10 mol%),in toluene(1.0 mL),18 h,60°C,argon.dr=the ratio of trans/cis.
We next explored the generality of thecis-products formation system(Scheme 3).To our delight,introduction of halogen-,MeO-,CF3-,CN-,Me2N-group into theparaposition of theβ-phenyl ring in the chalcone and other chalcones afforded the desiredcisproducts with excellent yield and diastereoselective ratio(cis-3aa tocis-3ea,63%−99%yield,dr=1:8 to 1:>20).The configuration ofcis-3ac was determined by X-ray crystallography(CCDC:2116931).Only starting materials were recovered with the NO2group substituted enone(cis-3ag).While,when the CF3-substituted enone was employed,the[4+2]cycloaddition productcis-3ia was obtained in 54%yield with low diastereoselectivity(dr=1:3).
The synthetically useful of this[4+2]annulation system was briefly investigated(Scheme 4).The coupling of 1a and 2aa was readily scaled up to 2.0 mmol,affording the producttrans-3aa in excellent yield and diastereoselectivity.Similarly,theβ-CF3substituted enone 2ia could also be scale up to 2.0 mmol(Scheme 4A).The ester 4 was obtained in 61%yield through Baeyer-Villiger oxidation oftrans-3aa.Wittig reaction oftrans-3aa gave the olefination product 5 in 95%yield.These two transformations with the retention of thetransconfiguration.Interestingly,when the CF3-substituted dihydrophenanthrenetrans-3ia was treated withtBuOK,CF2H-substituted phenanthrene 6(37%yield)and CF3-substituted phenanthrene 7(50%yield)were obtained in one pot under the air atmosphere(Scheme 4B).
We initially realized the enantioselective variant of this C–C activation system of biphenylene usingβ-CF3substituted enone 2ia as coupling partner(Scheme 5).With the chiral NHC ligand L1,the desired[4+2]annulation producttrans-3ia was obtained in 43%yield and 50%ee.
To determine the kinetic and thermodynamic control property of the reaction,we studied the isomerization of the two diastereoisomers of products(Scheme 6).Whencis-3aa was added to thetrans-3aa formation conditions(Reaction conditions A),thecis-3aa isomerized totrans-3aa with>20:1drvalue(Scheme 6A).Whiletran-3aa could not isomerize tocis-3aa under thecis-products formation conditions(Reaction conditions B)(Scheme 6B).These results indicated thetrans-3aa would be the thermodynamic product,andcis-3aa would be the kinetic product.
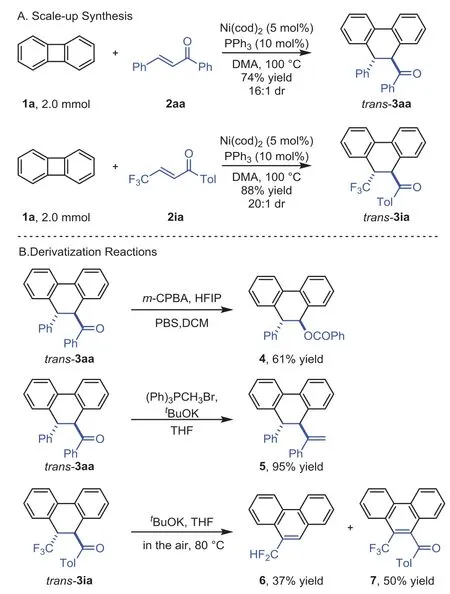
Scheme 4.Scale-up synthesis and derivatization.
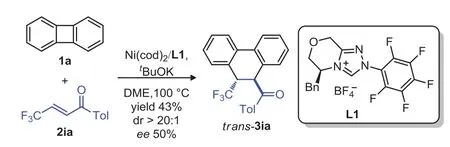
Scheme 5.Enantioselective coupling system.
On the basis of previous reports on the transition-metal catalyzed ring-opening of biphenylenes[32–51],a plausible pathway is proposed(Scheme 7).Oxidative addition of biphenylene 1a to Ni(0)afforded the intermediate A.The double bond of enone coordinated to the nickel center,which is proposed to undergo Ni-C insertion into the C=C bond of 2aa to generate the intermediate B.The intermediate B could isomerize to intermediate B’,followed by reductive elimination to give the kinetic productcis-3aa in toluene solvent and regenerated the catalyst.The high steric hindrance for thecis-isomer might benefit the reductive elimination process from the intermediate B in the toluene solvent[58–60].Thecis-3aa could isomerized to thermodynamic producttrans-3aa in DMA solvent(pathway A).Right now,we could not exclude the pathway of direct generation oftrans-3aa from the intermediate B in DMA solvent(pathway B).
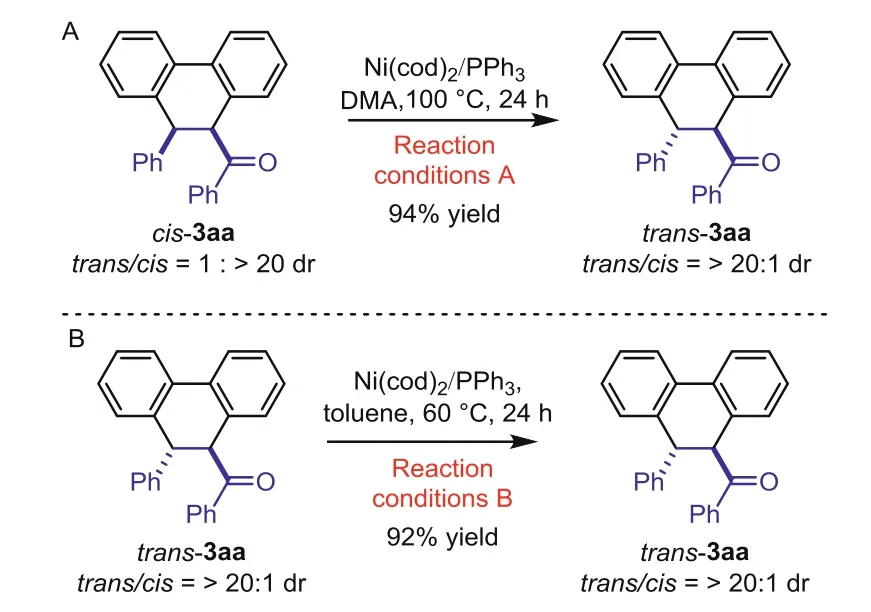
Scheme 6.Isomerization of the product.
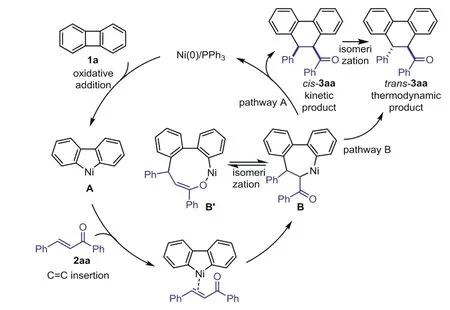
Scheme 7.Proposed mechanism.
In summary,we have realized Ni(0)-catalyzed diastereodivergent[4+2]annulation of biphenylene with enonesviaC–C bond activation.This reaction allows efficient access to the two diastereoisomers of the annulation products from the same starting materials just changing the reaction conditions.This solventcontrolled reaction proceeded with highly diastereoselectivity for both isomers(>20:1dr)and the C=C bond formally insertion chemoselectively in this cycloaddition.This intermolecular coupling system would be a straightforward strategy for the synthesis of 9,10-dihydrophenanthrene derivatives.
Declaration of competing interest
The authors declare no competing financial interests.
Acknowledgments
This work is supported by the National Natural Science Foundation of China(Nos.21801067,U1804283,21801066),the Central Plains Scholars and Scientists Studio Fund(No.2018002),and the Project funded by the Natural Science Foundation of Henan(Nos.202300410225,212300410181,222102310562 and 201901023)and China Postdoctoral Science Foundation(Nos.2020T130176 and 2020M682306).We also thank the financial support from Henan Key Laboratory of Organic Functional Molecules and Drug Innovation.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.02.079.
 Chinese Chemical Letters2022年12期
Chinese Chemical Letters2022年12期
- Chinese Chemical Letters的其它文章
- Diverse strategic approaches en route to Taxol total synthesis
- Recent advances in gold-complex and chiral organocatalyst cooperative catalysis for asymmetric alkyne functionalization
- Unmodified methodologies in target discovery for small molecule drugs:A rising star
- Recent advances in single-crystalline two-dimensional polymers:Synthesis,characterization and challenges
- Environmental applications of graphene oxide composite membranes
- Recent advances in the application of metal organic frameworks using in advanced oxidation progresses for pollutants degradation