
单细胞核酸编码扩增成像分析
2023-01-25 05:34赵雪琪宋大千赵永席
高等学校化学学报 2022年12期
赵雪琪,赵 越,薛 静,白 敏,陈 锋,孙 颖,宋大千,赵永席
(1.吉林大学化学学院,吉林省光谱分析仪器工程技术研究中心,长春 130012;2.西安交通大学生命科学与技术学院,生命分析化学与仪器研究所,生物医学信息工程教育部重点实验室,西安 710049)
细胞成像技术的革新不断推动生命科学的发展,通过成像实时清晰地观测细胞内及胞外微环境的细微变化,对于细胞分析十分重要.单细胞成像分析可提供不同细胞个体中分子的含量与分布信息,不仅在分析胞内分子结构、分子间反应、细胞间差异以及相互作用等方面应用广泛,还可以通过多组学联合分析,解析不同类型肿瘤细胞的耐药性差异,从而实现临床个体化精准治疗[1].在单细胞成像中,细胞中的物质变化需要通过探针作为媒介来实现信号的输出.其中,单细胞荧光成像由于具有荧光信号灵敏度高、特异性好及易于检测等优点,已经成为生命分析领域的研究热点[2].
目前,单细胞荧光成像的目标物已覆盖核酸表观修饰[3]、细胞酶活性[4]、蛋白质间相互作用[5]及胞内外小分子动态变化[6]等很多方面.其中,发展较为成熟的检测方法有荧光重组蛋白[7]、有机小分子荧光探针[8,9]及荧光核酸探针[10]等.荧光重组蛋白是由荧光蛋白与目标蛋白基因融合后再表达,具有很好的特异性,但蛋白体积较大且易发生荧光漂白.与之相比,有机小分子荧光探针的体积更小且荧光强度更高.然而,有机染料的膜通透性较差且容易发生非特异性吸附[11].
核酸荧光探针[12]具有可程序化设计、易修饰活性功能基团和可扩增放大检测信号等特点,在检测痕量目标物方面具有很大的应用潜力.由于荧光分子波谱重叠,常见方法大多受到检测通道种类的限制,检出限和灵敏度很难达到理想状态[13,14].然而,细胞内目标物种类多、丰度低,同源组分结构相似,分子邻近距离短.因此,如何实现单细胞内多组分标志物的精准识别与高灵敏定量分析,已经成为生命分析领域面临的重大挑战之一,核酸编码技术由此进入大众视野.
核酸编码技术最早于20世纪90年代被提出[15],在后期的不断完善和发展中,已被拓展应用于文库筛选[16,17]、测序[18]及成像[19,20]等方面.脱氧核糖核酸(Deoxyribonucleic acid,DNA)条码具有结构稳定、数目多及易于制备等特点[21],已成为核酸编码中应用最广泛的工具之一.针对不同目标物的特征,可通过不同的分子反应使其携带DNA条码,再通过这些条码上不同的荧光信号来区分目标物[22,23].由于单一条码分子的荧光强度难以直接检出,可联合核酸扩增技术实现信号放大[24].近年来,等温扩增以其温和的反应条件、快速有效的扩增和简单的操作被广泛用于生物成像分析中[25].在等温扩增中,常用的反应有滚环扩增反应(Rolling circle amplification,RCA)、杂交链式反应(Hybridization chain reaction,HCR)、引物交换反应(Primer exchange reaction,PER)和催化发夹组装反应(Catalyzed hairpin assembly,CHA)等.其中,RCA是环状探针和部分序列互补配对的引物探针杂交后,在聚合酶的作用下延伸引物并生成重复序列的过程.通常与单分子荧光原位杂交技术(Single-molecule fluorescentin situhybridization,smFISH)联合使用,杂交与重复序列互补的荧光探针来产生可检测的荧光信号点.PER是由单链引物和发夹结构的DNA在聚合酶的作用下反复进行链置换反应,从而将引物末端延长出重复序列的过程,可通过连续杂交信号探针放大检测信号.HCR是一对部分互补的发夹序列在被单链核酸杂交触发后,通过链置换反应不断交替打开更多发夹并生成长链DNA的过程.CHA与HCR类似,但两对发夹互补后形成的双链结构更稳定,从而使触发反应的单链核酸恢复游离状态,继续引发下一组发夹发生反应,生成大量的短链DNA.与RCA和PER相比,HCR和CHA不需要酶介导,但序列的设计较为复杂[25~27].因此,联合核酸编码与扩增技术,通过特异性识别转化分子特征为独特DNA条码的扩增,可实现特异、灵敏的单细胞多组分检测,对于当前生物和临床医学研究都有重大意义.
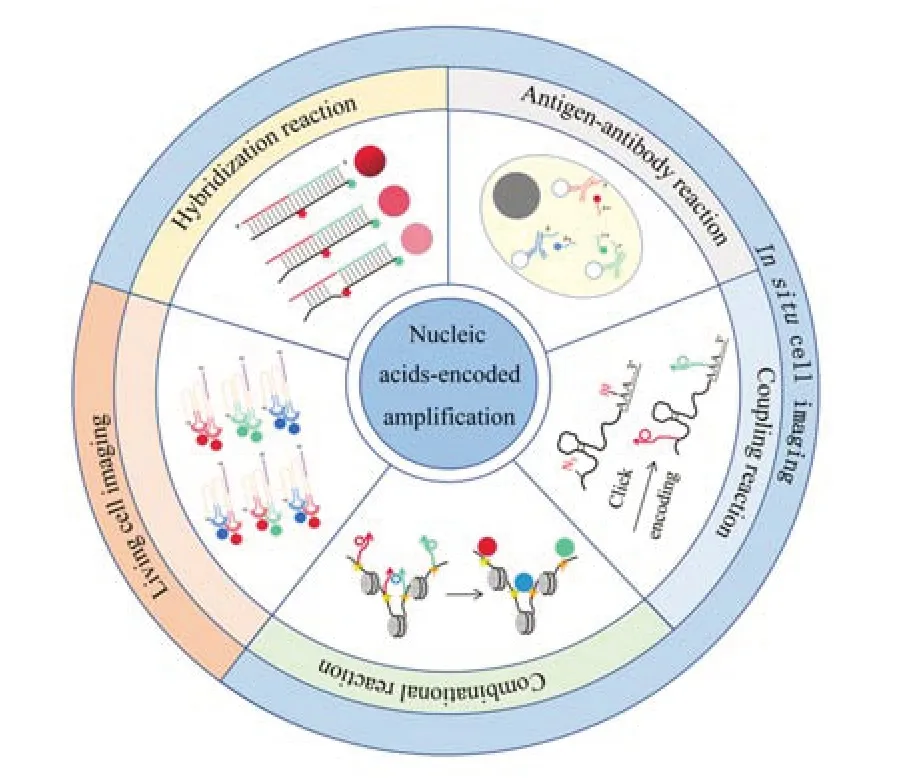
Fig.1 Progress in nucleic acids-encoded amplification for single-cell imaging
本文介绍了单细胞核酸编码扩增成像分析的最新进展,结合本课题组近期的研究成果,根据不同的编码原理从原位和活细胞角度对细胞内的核酸编码扩增成像方法进行介绍(图1),并对该技术的应用前景进行展望与分析.
1 原位核酸编码扩增成像
核酸编码扩增是将核酸编码与等温扩增结合从而实现多目标物识别转化检测的一种方法.其中最重要的一步就是通过化学反应给多种目标物标记上不同的DNA条码.目前常用于原位细胞成像的核酸编码方式主要基于杂交反应、化学偶联反应、抗原-抗体免疫反应与组合反应等,可与等温扩增反应结合,实现后续的信号放大.

Fig.2 Schematic of MERFISH[37]
1.1 基于杂交反应的编码原理
基于碱基互补配对原则的杂交反应是碱基相关反应中最常见的一种,也是核酸探针进行检测、成像和编码的基础.在核酸探针上修饰同位素[28]、生物素[29]或荧光基团[30]等可以对目标物进行成像和分析.对于已知序列,可以设计互补的核酸探针直接进行杂交反应编码[31,32];对于未知序列,则需要先连接上带有已知序列的通用接头再进行编码.smFISH是杂交编码成像的常用方法,通过荧光基团修饰的短核酸探针与目标物上不同的序列区域杂交,使目标物修饰上多个荧光分子,从而放大信号,实现单分子荧光成像[33,34].由于荧光基团种类的限制,smFISH很难成像多种目标物[35,36].
Zhuang等[37]提出的多重纠错荧光原位杂交(Multiplexed error-robust FISH,MERFISH)技术利用FISH组合标记和能够纠错的编码方法进行单细胞中核糖核酸(Ribonucleic acid,RNA)的成像,解决了smFISH在单个细胞中同时成像的RNA种类数量受限的问题,实现了空间分辨的单细胞转录组分析.针对特定目标RNA序列可设计连续杂交不同区域的多个编码探针,放大检测信号.编码探针由一段杂交序列和两段读出序列组成,每轮杂交时加入一种与某段读出序列互补的荧光探针,有荧光信号的记为“1”,没有荧光信号的记为“0”(图2).经过N轮杂交后,形成一段含有N个字节的二进制码,再通过汉明距离降低编码错误率,最终可以通过14轮杂交实现单细胞中1001种RNA的成像[图3(A)].由于FISH探针存在脱靶问题,在增加编码轮数、成像更短的RNA以及成像组织样本时,会产生脱靶背景[38].研究人员发现细胞内非特异性结合脱靶荧光探针的主要物质是蛋白质,所以选择将样本嵌入聚丙烯酰胺中以减小脱靶背景.样本内的RNA锚定在聚丙烯酰胺基质上,随后去除蛋白质,再进行MERFISH.实验结果表明,该方法在蓝绿光谱范围内使得MERFISH成像从2个通道扩展到4个通道,并且性能没有降低.因此减少了杂交编码的轮数,缩短了检测时间,提高了MERFISH的通量[38].基于MERFISH原理,该课题组发展了一系列应用.在扫描基因变异文库中[39],为每个含有待测遗传变异的质粒随机地分配一个条形码,使得每个细胞只检测一种高丰度的被编码的RNA,多轮荧光成像后进行信号的汇总,这样更亮的信号就有更低的错误率.通过这种方法识别了更亮更稳定的荧光蛋白YFAST中的60000个变异位点.此外,Zhuang等[40]还利用MERFISH建立一个分子级和空间分辨的小鼠初级运动皮层细胞图谱[图3(B)].在MERFISH基础上,他们结合分枝DNA(branched DNA,bDNA)扩增进行信号放大[41].在MERFISH第一步杂交完成后,加入一端与读出条码互补、另一端带有多段重复序列的初级信号放大条码进行第一次放大.二级信号放大条码的结构与初级信号放大条码一样,一端与初级信号放大条码互补杂交,另一端与多个荧光探针杂交,使MERFISH的信号进行两轮放大,提高效率.

Fig.3 Data of application of MERFISH
Cai等[42]提出了连续单分子荧光原位杂交(Sequential FISH,seqFISH)的方法,对固定细胞中的信使RNA(Message RNA,mRNA)进行连续探针杂交,每轮探针带有不同颜色的荧光团,在一轮杂交结束后,通过DNA酶降解探针,再进行下一轮杂交[图4(A)].因为细胞是固定的,所以对应每个mRNA的荧光点在每轮成像中都保持在原位,由此产生一种独特的荧光序列条码,实现mRNA的成像.由于smFISH在组织成像中较难实现,当前发展的基于seqFISH的方法都是细胞成像.为了实现复杂组织的成像,他们从信号放大和高效的误差修正两方面对seqFISH进行改进,对海马的结构组织进行单细胞分辨率的解析[43].该方法采用单分子杂交链式反应(Single-molecule HCR,smHCR)进行信号扩增,多个编码探针杂交到mRNA上后,探针上突出的单链部分通过互补杂交打开带有荧光-猝灭基团的发夹1,发夹1再用同样的方式打开发夹2,不断循环生成带有很多荧光基团的DNA长链聚合物.该smHCR产物可以被DNA酶消化,并且mRNA在脑组织切片中可以重新杂交,从而实现HCR-seqFISH[图4(B)].
Deisseroth等[44]提出了一种能在完整组织中进行RNA原位测序的方法,命名为空间分辨转录本扩增示值图谱分析(Spatially-resolved transcript amplicon readout mapping,STARmap).他们将含有不同DNA条码的挂锁探针杂交到mRNA上,在RCA过程中加入一部分氨基修饰的碱基,最终得到含有多个相同条码和氨基修饰的RCA产物.丙烯酸N-羟基琥珀酰亚胺酯与氨基反应,得到丙烯酰胺基修饰的RCA产物,随后与丙烯酰胺单体共聚形成水凝胶-DNA扩增子网络结构.该结构由共价键连接,可以在后续去除蛋白和多轮探针清洗中保持稳定.DNA条码由5个随机碱基组成,形成1024个不同的条码.只有当读出探针和荧光探针与DNA条码完全杂交时,才能稳定连接并产生荧光信号.每轮识别一个碱基,读出探针的碱基数逐个递增,荧光探针的碱基数逐个递减[图4(C)].在一轮识别结束后,加入甲酰胺去除核酸探针,消除测序过程中错误的累积.最后通过与相应的基因进行序列比对,STARmap实现了小鼠大脑切片上单细胞分辨率的160~1020个基因的检测.
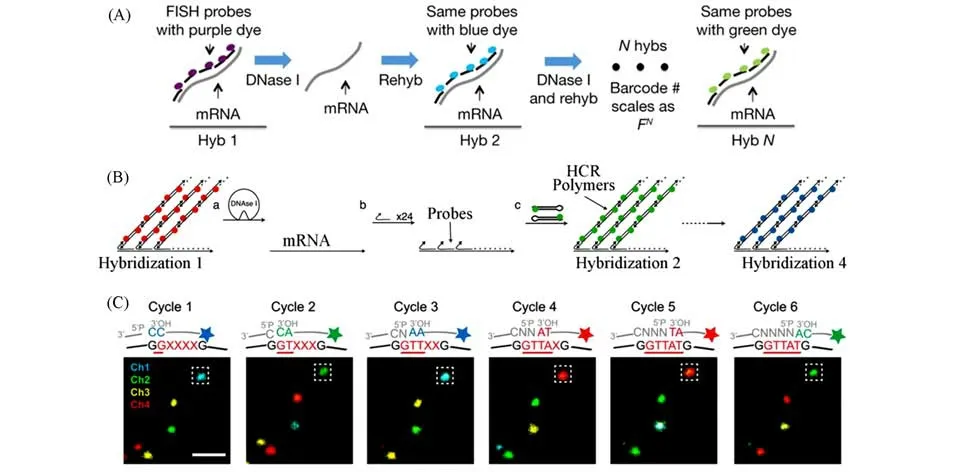
Fig.4 Schematic of hybridization encoded amplification methods
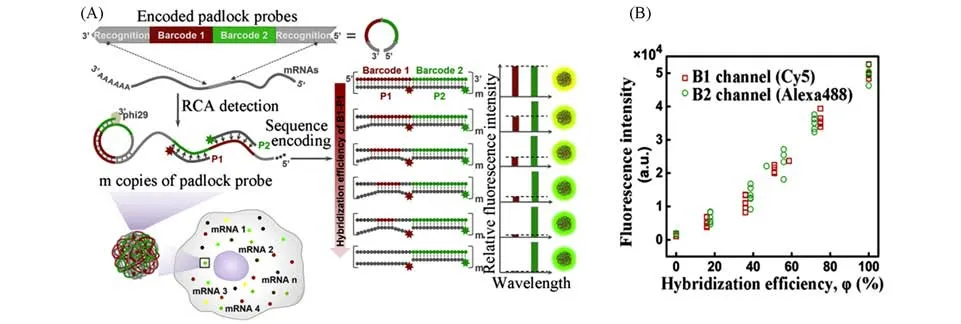
Fig.5 Schematic and data of SeqEA[46]
Yin等[45]提出了一种基于FISH的信号放大方法,命名为基于交换反应的荧光原位杂交信号放大法(Signal amplification by exchange reaction-FISH,SABER-FISH).DNA条码由信号识别条码和信号放大条码两部分组成,信号识别条码和目标物互补杂交,信号放大条码预先在体外通过PER合成含有重复序列的长链.当DNA条码和目标物杂交后,每轮加入一种与信号放大条码互补的荧光探针进行快速杂交成像,然后用低离子强度缓冲液迅速洗脱探针,再进行下一轮杂交,以实现多轮编码扩增成像.
Li等[46]提出了一种荧光编码扩增方法——核酸序列编码扩增(Sequence-encoded amplicon,SeqEA),利用目标RNA介导环形识别探针环化并触发RCA信号放大,通过热力学调控RCA产物与荧光探针的杂交效率实现荧光信号的改变,即可对目标物进行荧光编码.与传统RCA荧光成像不同的是,SeqEA中RCA的挂锁探针分为两段条码,分别对应带有红色和绿色荧光基团的两种荧光探针.改变挂锁探针上与荧光探针对应段的碱基序列,就会产生不能与荧光探针完全配对杂交的RCA产物,从而降低荧光探针的杂交效率,使荧光强度降低(图5).通过这种方法设计了36个双色荧光条码,当使用更多颜色的荧光基团时,SeqEA的编码能力可以成倍提高.
基于FISH发展的多轮杂交编码成像实现了大量目标物的检测,并在后续发展中不断完善,对于构建文库和生物体的全局分析起到了非常重大的作用.但是杂交编码原理仅能识别序列差异,无法分辨碱基修饰基团、核酸空间邻近关系等非序列特征信息.

Fig.6 Schematic and data of Clicker-FISH[53]
1.2 基于化学偶联反应的编码原理
化学偶联反应编码通常是将化学反应活性基团标记到核酸探针上,通过化学基团与目标物之间的特定化学反应使目标物标记上不同的DNA条码.DNA编码的小分子文库筛选中提供了很多修饰方法和化学反应[16,47,48],其中,点击化学反应因其生物相容性好、操作简单、反应条件温和而广泛应用于细胞成像中[49~52].
在本课题组的研究工作[53]中,点击化学反应编码滚环扩增的荧光原位杂交成像(Click-encoded rolling FISH,Clicker-FISH)已被用于单细胞RNA多聚腺苷酸化及结构的可视化成像.胞内RNA会在其加工过程中产生多聚腺苷酸(Polyadenylic acid,polyA)尾巴,部分RNA还会折叠成部分单链、部分双链的茎环结构.成像可获取PolyA、单链RNA(Single-stranded RNA,ssRNA)和双链RNA(Double-stranded RNA,dsRNA,此方法中即茎环结构)等加工与结构信息,揭示其亚细胞时空分布,对于解析细胞状态具有重要意义.结合活细胞代谢标记和固定细胞化学修饰标记方法,使3种目标物分别标记上不同的化学基团,其标记方法分别对应以下3种反应机理:(1)2-炔基腺苷(2-Ethynyl-adenosine,2-EA)在活细胞代谢中被聚合到polyA尾巴上;(2)细胞固定后,叠氮修饰的RNA酰化试剂(Azide-modified 2-methylnicotinic acid imidazolide,NAI-N3)特异性与单链暴露的2-OH基团发生酰化反应;(3)在365 nm紫外光照射条件下,通过环辛烯功能化的补骨脂素衍生物(Trans-cyclooctene psoralen,TCO-Pso)标记RNA双链区域,与双链RNA上的嘧啶对交错反应[图6(A)].随后,铜催化的叠氮与炔烃、无铜的叠氮与二苯并环辛炔(Dibenzocyclooctyne,DBCO)和烯烃与四嗪(Tetrazine,Tz)3种点击化学反应分别将3种不同的DNA条码标记到目标物上.对3种DNA条码进行RCA后,杂交荧光探针进行成像[图6(B)].基于此原理,该方法在增加DNA条码的情况下也可以实现RNA及相关蛋白的可视化成像,与单细胞转录组成像方法进行整合后,在研究转录组结构及序列成像方面也有很大的应用潜力.
此外,本课题组还通过偶联编码进行了细胞内表观修饰的识别与成像.Zhao等[54]提出了一种利用生物正交的化学标记成像单细胞内两种氧化胸腺嘧啶的方法.他们对5-羟甲基尿嘧啶[5-(Hydroxymethyl)uracil,5hmU]和5-甲酰基尿嘧啶(5-Formyluracil,5fU)偶联编码标记后扩增,结合荧光成像和测序,实现了对基因组中两种氧化胸腺嘧啶的量化和精确绘制.在5-羟甲基尿嘧啶磷酸激酶(5hmU DNA kinase,5-HMUDK)作用下,γ磷酸炔基化的三磷酸腺苷类似物(Adenosine triphosphate-γ-alkyne,ATP-γalkyne)通过化学酶标法特异性识别5hmU并标记上炔基,叠氮修饰的DNA条码通过点击化学反应连接上炔基修饰的5hmU.随后,硼氢化钠将5fU还原成5hmU,用同样的方法给5fU标记上另一种DNA条码,对两个条码进行RCA和FISH成像,实现单细胞内两种修饰碱基的可视化定量.
除了点击化学反应,马来酰亚胺与巯基的偶联反应应用也很广泛.本课题组将微液滴凝胶与偶联核酸编码整合,提出了一种差异化成像单细胞内两种5-羟甲基嘧啶的方法,命名为单细胞微流凝胶液滴5hmU和5hmC差异可视化(Single-cell 5hmU and 5hmC differentiated visualization in microfluidic hydrogel droplets,sc5hmU/5hmC-microgel)[23].在液滴捕获单细胞后,利用凝胶结构固定胞内的大分子核酸.γ磷酸巯基化的三磷酸腺苷类似物(Adenosine-5-O-thiotriphosphate,ATP-γ-S)在5-HMUDK作用下将5hmU特异性标记上巯基,随后T4噬菌体β-葡萄糖基转移酶(T4 phageβ-glucosyltransferase,β-GT)将5-羟甲基胞嘧啶(5-Hydroxymethylcytosine,5hmC)特异性标记上叠氮葡萄糖.再通过化学偶联反应使马来酰亚胺和DBCO修饰的DNA条码分别标记到5hmU和5hmC位点,经RCA后成像两种碱基的位置(图7).该方法实现了对于相似组分的编码成像,并结合微流液滴进行单细胞分析,扩大了微流芯片和核酸编码扩增成像的应用.
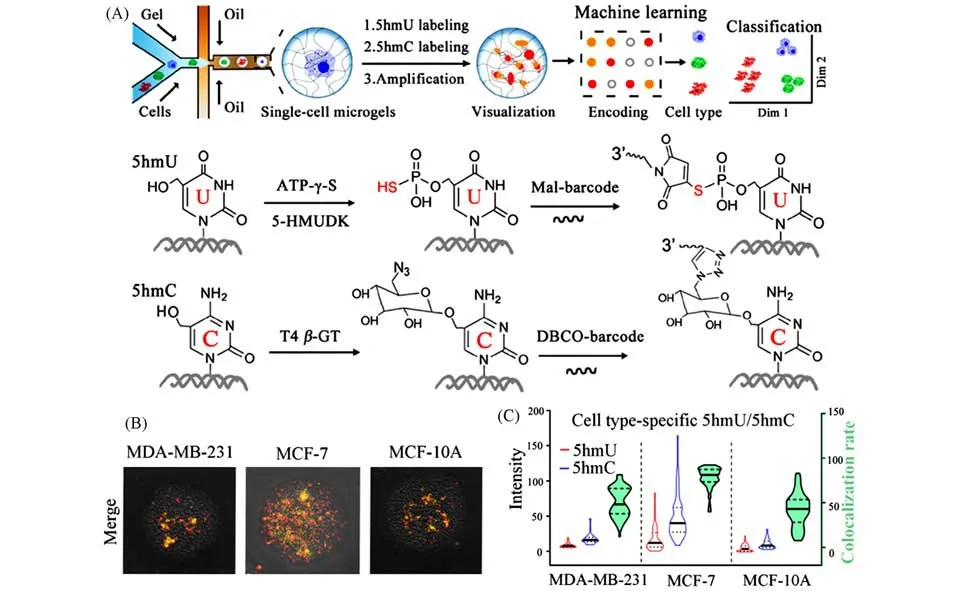
Fig.7 Schematic and data of sc5hmU/5hmC-microgel[23]
与利用杂交反应进行条码标记的方式相比,化学偶联反应可以对未知序列以及不能直接杂交核酸探针的目标物(如修饰碱基、蛋白和双链核酸等)进行DNA条码的标记,扩大了核酸编码的应用范围.但是目前反应条件温和、高效、生物相容性好的化学偶联反应种类有限,如何拓展更多的反应也是需要关注的方面之一.
1.3 基于抗原-抗体免疫反应的编码原理
抗原-抗体免疫反应在生物分析检测中已经发展成熟,在此基础上的三明治信号放大方法也有很多种,如抗体-抗原-抗体、抗体-抗体-核酸等.由于抗体本身具有识别功能且种类很多,因此将抗体与DNA条码通过通用的偶联反应结合在一起形成抗体-核酸编码探针可以扩大目标物的识别范围[55].
Yin等[56]利用同时带有N-羟基琥珀酰亚胺(N-hydroxysuccinimide,NHS)酯基和马来酰亚胺基团的化学物质将抗体和DNA条码连接到一起形成抗体-核酸探针.NHS酯基和马来酰亚胺基团分别与赖氨酸上残留的氨基和巯基修饰的DNA条码发生反应[图8(A)].抗体-核酸探针可以直接识别目标物,也可以作为二级抗体进行信号放大.作者还将Tz修饰的DNA条码和反式环辛烯(Trans-cyclooctene,TCO)修饰的纳米抗体通过点击化学反应结合到一起.纳米抗体尺寸可以达到~1.5 nm×2.5 nm,在很大程度上提高了荧光信号空间位置的准确性.此外,他们还提出了一种快速洗脱成像的方法[57],也应用在SABER-FISH中.修饰不同DNA条码的抗体-核酸探针识别目标物后,每轮加入一种荧光探针杂交,再用低离子强度缓冲液迅速洗脱探针,进行下一轮杂交,原则上可以循环无限次.由于每轮反应时间很短,探针杂交不够稳定,因此容易被洗掉,但也会带来荧光信号弱且不稳定的问题.
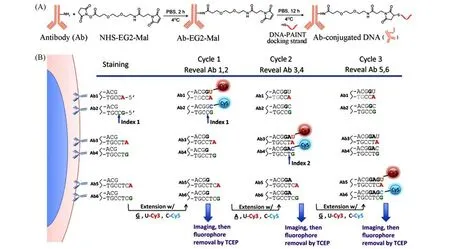
Fig.8 Schematic of antigen-antibody encoding methods
Nolan等[58]提出了一种高度多路成像的方法索引共检测(Co-detection by indexing,CODEX),通过比较正常小鼠与患免疫疾病小鼠的脾脏,观察脾细胞的相互作用动力学.将5′突出的双链DNA条码修饰到抗体上,每组抗体上5′突出的长度逐个递增,称为索引(Index)碱基,每轮增加的Index碱基都和上一轮不同.每组包含两个抗体,同组抗体上的两个DNA条码只有5′端的第一个碱基不同,作为信号碱基,并且这两个碱基不会出现在Index碱基里.每轮成像加入3种碱基,分别与带有荧光团的两种信号碱基和一种Index碱基互补,成像后用三(2-羧乙基)膦盐酸盐[Tris(2-carboxyethyl)phosphine hydrochloride,TCEP]洗掉荧光基团,再进行下一轮成像[图8(B)].虽然理论上两种Index碱基可循环使用,能检测无限个目标物,但由于碱基数量限制,每轮只能成像两种目标物,难以实现高通量检测.
为了进一步放大荧光信号,Luo等[59]将抗原-抗体免疫反应与HCR结合在一起,称为免疫响应杂交链式反应(Immunosignal HCR,isHCR)方法.该方法将免疫荧光信号放大2~3个数量级.一级抗体识别抗原后,生物素标记的二级抗体与一级抗体结合,链霉亲和素修饰的DNA探针与生物素反应后标记在目标物上,HCR放大信号后进行荧光成像.虽然和荧光基团直接标记相比,isHCR实现了荧光信号增强,但是受限于二级抗体的数量,能检测的目标物有限.如果在HCR的过程中结合二次编码扩增,就能扩大检测范围.
Liu等[60]提出了荧光寡核苷酸联免疫吸附斑点法(Fluorescence-based oligo-linked immunospot,FOLISPOT),通过酶联免疫吸附斑点法(Enzyme-linked immunospot,ELISpot)和两轮PER放大信号方法的结合,将检测限降为ELISpot检测限的2%,并实现了多轮检测.捕获抗体、抗原和检测抗体形成三明治结构,检测抗体和DNA条码通过马来酰亚胺-聚乙二醇-琥珀酰亚胺酯交联剂连接到一起.随后,通过第一轮PER延长与抗体上DNA条码互补的链,该链又作为第二轮PER的底物,生成大量可与荧光探针互补杂交的单链,实现荧光信号的放大.固定在孔板上的捕获抗体提供了洗涤界面,使得反应体系可以进行多轮荧光成像,即修饰相同荧光分子的不同核酸探针可以在不同轮次内成像.本方法每轮成像只用了两种荧光分子,如果增加每轮中荧光分子的种类,可以在多轮洗涤中识别更多目标物[图9(A)].
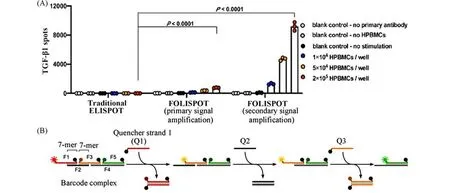
Fig.9 Data and schematic of FOLISPOT and CCFB
Kashida等[61]提出了颜色变换荧光条码(Color-changing fluorescent barcode,CCFB)方法,通过多肽与肌动蛋白结合或抗原抗体结合使目标蛋白带上组合条码,随后进行多轮链置换反应对目标多轮成像,形成不同顺序和颜色的荧光编码.与之前方法不同的是,荧光探针用D-苏氨醇非环核酸(AcyclicD-threoninol nucleic acids,D-aTNA)代替DNA.D-aTNA自身能形成非常稳定的双链,但不能与DNA或RNA形成稳定的双链结构.加入D-aTNA单链猝灭探针后,与荧光探针通过链置换形成的荧光猝灭双链产物结构稳定,不会被其它DNA单链置换,因此不需要在每轮识别后洗涤,缩短了每轮检测的时间.此外,与DNA荧光探针相比,D-aTNA荧光探针也有更高的猝灭效率和信噪比[62].不同组合的探针核酸序列是相同的,调整所带荧光基团的顺序即可实现不同的编码,因此链置换的反应是通用的,避免了大量不同核酸链的引入[图9(B)].巯基修饰的D-aTNA组合探针通过交联剂连接到抗体或鬼笔环肽上,进行抗原或纤维状肌动蛋白的识别.由于重复的荧光基团也有顺序差异,因此增加链置换的次数可以在有限荧光基团种类的基础上实现更多种目标物的检测.
基于抗体识别的核酸编码扩增,可检测的目标物种类更丰富、特异性更强.但是抗体尺寸较大且二抗种类有限,在识别过程中容易造成位置信息不准确.而纳米抗体虽然尺寸缩小很多,但是制备过程较复杂.
1.4 基于组合反应的编码原理
为了获取多种目标物间的位置关系、相互作用情况等多层次分子信息,本课题组开发了基于多种分子逐级反应的组合编码原理.在偶联反应编码的基础上,Zhao等[63]利用1,3-茚二酮(1,3-Indandione,AI)叠氮衍生物将5-甲酰基胞嘧啶(5-Formylcytosine,5fC)特异性标记上叠氮基团,并与DBCO修饰的DNA条码发生点击化学反应,同时采用sc5hmU/5hmC-microgel方法标记5hmC,由此可为两种目标物分别标记上部分序列互补的单链DNA条码.进一步开发邻近连接反应编码原理,通过邻近分子激活新的DNA条码,可在获得不同分子信息的同时获得邻近分子的关联信息.挂锁探针和两种DNA条码分别有一段互补序列,只有当两种条码足够近,即5hmC和5fC位点相邻时,挂锁探针才能与两个条码发生邻近连接反应[64,65]被激活,在RCA后进行荧光成像.在识别过程中,邻近连接的挂锁探针标记后,再用另外两种挂锁探针对剩余的5hmC和5fC位点进行标记,避免了先占用邻近连接的位点.
针对更加复杂的生物分子的空间邻近关系,本课题组发展了细胞大分子锚定的DNA步移索引成像方法(Cellular macromolecules-tethered DNA walking indexing,Cell-TALKING)[22]方法,通过组合偶联反应编码、抗原-抗体免疫反应编码和邻近连接反应编码成像组蛋白与其周围5hmC,5hmU和5fU的位置关系.在这4种目标物的标记方法中,组蛋白的条码通过抗原-抗体免疫反应标记,5hmC的标记方法与sc5hmU/5hmC-microgel方法[23]相同,5hmU和5fU的标记方法与生物正交的化学标记成像单细胞内两种氧化胸腺嘧啶的方法[54]相同.位于组蛋白上的中心条码和周围表观修饰的3种DNA条码有一段通用的杂交互补序列,称之为“邻近条码”.当组蛋白与第一个目标物邻近时,中心条码和第一个DNA条码发生杂交,并在聚合酶作用下延伸,互补形成一段带有切刻酶位点的序列.加入切刻酶后,互补链在作用位点处发生断裂,杂交长度变短,双链解旋,中心条码恢复一端游离状态,同时第一个DNA条码被标记上中心条码的互补序列.通过不断重复这个过程,组蛋白周围所有邻近的3种表观修饰位点都会被标记上中心条码,再通过RCA后成像其位置.CELL-TALKING的位置关系在已知修饰位点距离的DNA折纸上也进行了验证(图10).
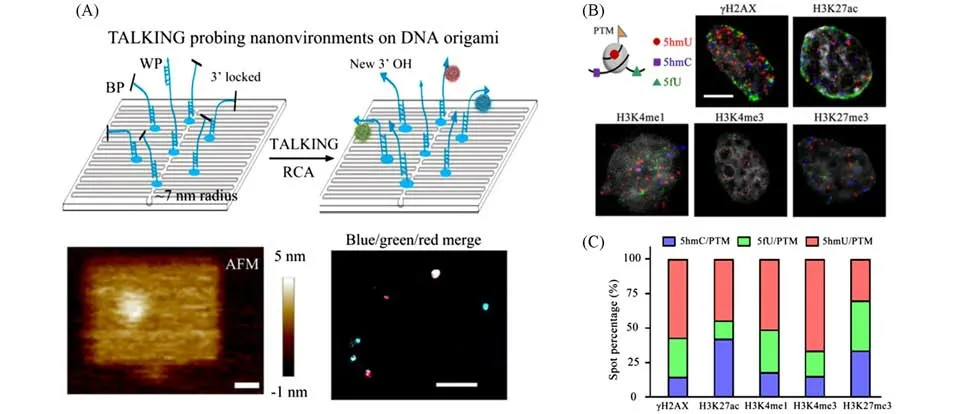
Fig.10 Schematic and data of Cell-TALKING[22]
Raj等[49]联合杂交反应和化学偶联反应,提出了一种放大FISH信号的检测方法,即点击化学反应放大荧光原位杂交法(Click-amplifying FISH,clampFISH).他们将一级挂锁探针的两个末端分别修饰炔基和叠氮基团,挂锁探针与目标物mRNA杂交后,在铜催化下发生点击化学反应连接成环,降低了环直接杂交目标物时的非特异性吸附.与连接酶将碱基连接成环相比,点击化学反应连接提高了连接效率.冲洗后,带有荧光基团的二级挂锁探针通过同样的方法杂交到一级挂锁探针上.不断循环这个过程,每一级荧光探针都与上一级探针发生杂交-点击化学反应,直到荧光信号达到理想强度.
组合反应编码方法通常是先用一种编码方式使目标物标记上DNA条码,再通过二次编码使之前的DNA条码再被标记一个新的条码.多次编码对于逐步深入研究目标物之间的关系发挥了重大作用.
2 活细胞核酸编码扩增成像
由于细胞膜具有选择透过性,无法重复进行杂交清洗,因此在固定细胞中常用的smFISH和RCA等方法不能直接应用在活细胞中.目前常用的活细胞内成像的方法主要有3类:第一类是通过细胞膜的胞吞胞吐作用,将带有核酸探针的纳米材料运输进细胞内,在胞内激活后进行后续的反应[66~68];第二类是用和细胞膜类似的物质(如脂质体)包裹探针,通过膜融合将探针运输进细胞内[69~71];第三类是将质粒转染到细胞内,通过在细胞内表达检测所需的核酸或蛋白进行成像[72~74].
纳米粒子在大量运输核酸探针进入细胞方面有着显著优势,金纳米粒子和硅纳米粒子都是常用的纳米材料[75~77].Zhang等[78]制备了一种负载DNA的金纳米粒子3D水凝胶结构(Au nanoparticle DNA hydrogel,AuDH),包含3种发夹结构的脱氧核糖核酶(DNAzyme)、与之部分序列互补的DNA荧光探针和激活的金属离子.当AuDH进入细胞内后,目标物微核糖核酸(microRNA,miRNA)通过链置换反应与DNA荧光探针杂交,打开AuDH,发夹DNAzyme和金属离子被释放出来.DNA荧光探针再次通过链置换反应打开DNAzyme的发夹结构,目标物miRNA被释放,继续打开下一个AuDH,实现第一次循环放大.与DNA荧光探针杂交的DNAzyme在金属离子的激活下,切断底物中的核糖核苷酸位点,使荧光探针的荧光团远离猝灭端,发出荧光信号.同时,DNAzyme与底物解旋后恢复游离状态,再识别下一个底物,实现第二次循环放大.AuDH负载了3套不同的miRNA检测系统,可以实现3种miRNA的同时成像[图11(A)].除了金纳米粒子外,Yao等[79]合成了负载染料的介孔二氧化硅纳米猝灭剂(Black hole quencher-doped mesoporoussilica nanoparticles,qMSNs).他们用目标miRNA互补链封装染料,将与甘油醛3-磷酸脱氢酶(Gyceraldehyde 3-phosphate dehydrogenase,GAPDH)mRNA互补的发夹荧光探针修饰在纳米粒子表面.miRNA与互补链结合后,染料被释放,发出荧光信号;同时,GAPDH的mRNA将发夹结构打开,使荧光基团远离猝灭基团,恢复荧光.该方法虽然结构设计相对简单,但没有循环放大信号的功能,在多检测和信号扩增上还有待进一步改善.Zhao等[66]设计了一种金纳米粒子负载的DNA信号放大系统,可以同时成像细胞内的miRNA和端粒酶.纳米粒子上带有两种包含氧桥鸟嘌呤(Oxoguanine,oG)损伤位点和荧光基团的DNA发夹以及两种未激活的DNA“开关”.纳米粒子进入细胞内后,miRNA与其对应开关的封锁链发生链置换反应,激活单链DNA开关.开关再通过链置换反应打开发夹,形成带有oG位点的DNA双链.同时,端粒酶在识别其对应开关后将末端延长从而激活,延长后的DNA单链开关同样通过链置换反应打开发夹并形成含有损伤位点的双链结构.细胞内的8-羟基鸟嘌呤DNA糖苷酶(Human 8-oxoguanine glycosylase 1,hOGG1)识别并切除oG,形成脱嘌呤嘧啶(Apurinic-Apyrimidinic,AP)位点,在体内碱基切除修复系统(Base-excision repair,BER)的作用下,双链解旋,释放出荧光信号和已激活的单链开关,进行下一轮的识别和放大.
由于纳米金类的纳米材料制备繁琐,且生物毒性存在争议,因此利用DNA组装的纳米结构在活细胞编码中也发挥着重要作用.Xiang等[80]设计了一种多色编码的DNA四面体纳米结构Tetrahedron nanostructure(TetrNano),用于多路检测细胞内miRNA.通过对4条单链进行热退火和连接,得到含有两个荧光猝灭发夹的四面体DNA纳米结构.MiRNA-21和miRNA-155分别打开两个发夹,使其恢复荧光,实现两种目标物的同时成像[图11(B),(C)].为了进一步放大信号以检测更低浓度的目标物,Yang等[81]组装了一种类似章鱼触手的纳米结构.两组CHA探针、AS1411适配体和生物素标记的DNA链组成了DNA三棱镜纳米结构(DNA triangular prism,DTP).3个DTP通过链霉亲和素组成Streptavidin DTP(SA-DTP).AS1411适配体可以帮助SA-DTP更顺利地进入细胞.当miRNA-21在第一个DTP上触发对应的CHA后,由于空间约束效应,重新释放的目标会快速传递到相邻的DTP上,触发其CHA,从而实现信号放大,miRNA-155同理[图12(A)].虽然DNA纳米结构的序列结构设计较复杂,但在生物相容性和可控性方面有着显著优势.
类膜结构的运输可以提高运输效率,同时避免复杂的组装过程.Zhao等[69]发现,通过脂质体将DNA回路所需的探针运输进细胞内后,细胞内的端粒酶会激活启动探针,开启回路.随后,两种RNA分别触发各自的链置换循环反应,放大信号[图12(B)].二进制编码原理同MERFISH.该方法利用细胞内熵驱动的多重DNA回路和信号编码,分析了细胞内端粒酶和两种RNA的关系.与常用的酶驱动扩增体系相比,无酶信号放大由于反应条件简单,避免了酶促反应中pH值和离子浓度的限制,在活细胞内效率更高.Dong等[82]通过肿瘤细胞膜囊泡包裹修饰在纳米粒子上的荧光探针和双链特异性核酸酶(Double-specific nucleases,DSN),对细胞内的3种miRNA进行成像.当miRNA与荧光探针结合形成双链后,会被DSN迅速识别并将DNA链水解,发出荧光信号.miRNA被释放后继续杂交下一条荧光探针,以此实现信号放大.
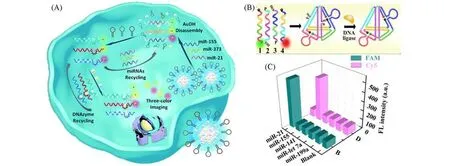
Fig.11 Schematic and data of nanoparticles and DNA nanostructures encoded amplification methods in live cells
Pederson等[83]提出了CRISPRainbow方法,可以在活细胞内利用可编程的规律间隔成簇短回文重复序列(Clustered Regularly Interspaced Short Palindromic Repeats,CRISPR)及其内切酶失活相关蛋白9(Dead CRISPR Associated Protein,dCas9)系统进行DNA标记成像.与之前改造规律间隔成簇短回文重复序列相关蛋白9(CRISPR Associated Protein9,Cas9)蛋白的方法不同,CRISPRainbow中结合荧光蛋白的对象为向导RNA(Single guide RNA,sgRNA).选用蓝色、绿色和红色的荧光蛋白,将带有与目标序列互补的sgRNA与含有6种组合荧光蛋白(3种单色和3种组合色)结合位点的适配体连接,构建的质粒在细胞内表达后,通过CRISPR-dCas9靶向结合目标物,进行荧光成像[图12(C)].在此基础上,该团队又提出了CRISPR-Sirius方法,将8组重复的MS2RNA适配体插入到sgRNA四环上,结合更多的荧光蛋白,进一步放大了荧光信号,可以检测更低拷贝数的目标物[84].
活细胞中条形码的表达水平受多种因素影响,可能干扰后续检测的准确度,因此,Elowitz等[85]开发了一种不依赖于条形码表达的高精度原位检测技术Zombie.将T7/SP6/T3噬菌体启动子驱动的DNA条码整合到基因组中,这些条码在细胞中由于缺乏相关酶而不会表达.细胞内的碱基编辑器或其它DNA修饰酶可以选择性地改变条形码序列,以增加条形码的多样性.细胞固定后,引入噬菌体的RNA聚合酶,使带有DNA条码的目标序列开始原位转录扩增,RNA转录本由此开始积累并扩散,再通过FISH进行成像分析.此外,Zombie还可以有效区分20个碱基对长度的DNA条形码上单个碱基的差异.在编码成像中既保留了细胞的空间位置信息,也提高了条码分析精确度.类似的方法也可以追踪体细胞突变的谱系信息.Cai等[86]提出了基于基因工程诱导突变与光学原位读取的细胞历史记录(Memory by engineered mutagenesis with opticalin situreadout,MEMOIR)方法,将CRISPR/Cas9和seq-FISH结合,编码后通过原位成像分析谱系遗传信息.条码由10段重复序列组成,在CRISPR/Cas9特异性切割下产生随机的条码缺失.每段条码还带有一段不被Cas系统识别的颜色条码,通过seqFISH共定位成像.Cas系统与seqFISH结合也可以实现细胞中染色质动态的可视化[87].荧光蛋白标记的dCas9在活细胞中靶向到目标位点,随后将细胞固定,通过seqFISH多轮成像.这类在活细胞中编码、固定细胞后成像的方法有效解决了活细胞中不能多轮洗涤探针的问题.
活细胞中核酸编码扩增受到细胞膜选择透过性、生物相容性、无界面等诸多限制,在多轮编码成像多种目标物方面还有很大的阻碍.目标物的编码方式和扩增方式都与固定细胞中的有所不同.如何在活细胞中实现成百上千甚至更多种目标物的编码成像,依然是需要继续研究的问题.

Fig.12 Structure of DNA nanostructures and circuits and schematic of CRISPRainbow
3 总结与展望
本文综合评述了单细胞核酸编码扩增成像分析的方法以及在生命科学领域的研究进展.通过核酸编码扩增的方式对现有方法进行了分类和总结,部分方法列于表1.核酸编码与信号扩增结合,不仅可用于基于杂交识别的核酸序列检测(如mRNA和miRNA),还拓展应用于核酸非序列特征(碱基修饰、空间邻近)以及蛋白等多层次、多水平目标物分析,极大扩展了单细胞荧光成像的目标物范围与应用场景.
核酸编码扩增利用化学反应特异性识别标记特定目标物,将相似结构分子间的微小化学结构差异转化为预先设计的DNA条码的扩增反应.进一步联合多轮扩增编码,即可同时特异、灵敏地检出上万种目标物,为细胞内多种物质联合分析提供了有力的工具,在全基因组的数据分析和生物组织谱图绘制中发挥重大的作用.
目前,单细胞核酸编码扩增成像还面临一些挑战.首先,缺少高通量、自动化的单细胞成像方法.目前组学级别的目标物编码成像大多依赖多轮杂交、清洗,步骤繁琐,耗时较长,检测通量受限,迫切需要发展快捷、简便、能满足规模化单细胞并行检测的成像方法与平台.其次,目标物标记条码的方式单一.除序列杂交与抗原-抗体免疫识别外,对于其它分子特征仍局限于点击化学等少数几种反应,发展反应效率高、反应条件温和的多类型生物正交反应,将为单细胞成像分析提供更多的分子工具.最后,由于细胞膜的选择透过性,外界物质进入细胞的方式与效率有限,信号方式单一,放大效率不足,多目标物检测难度极大.如何在活细胞中进行多轮编码,以获取细胞内物质随时间、空间的动态变化信息,是值得关注的重点问题.
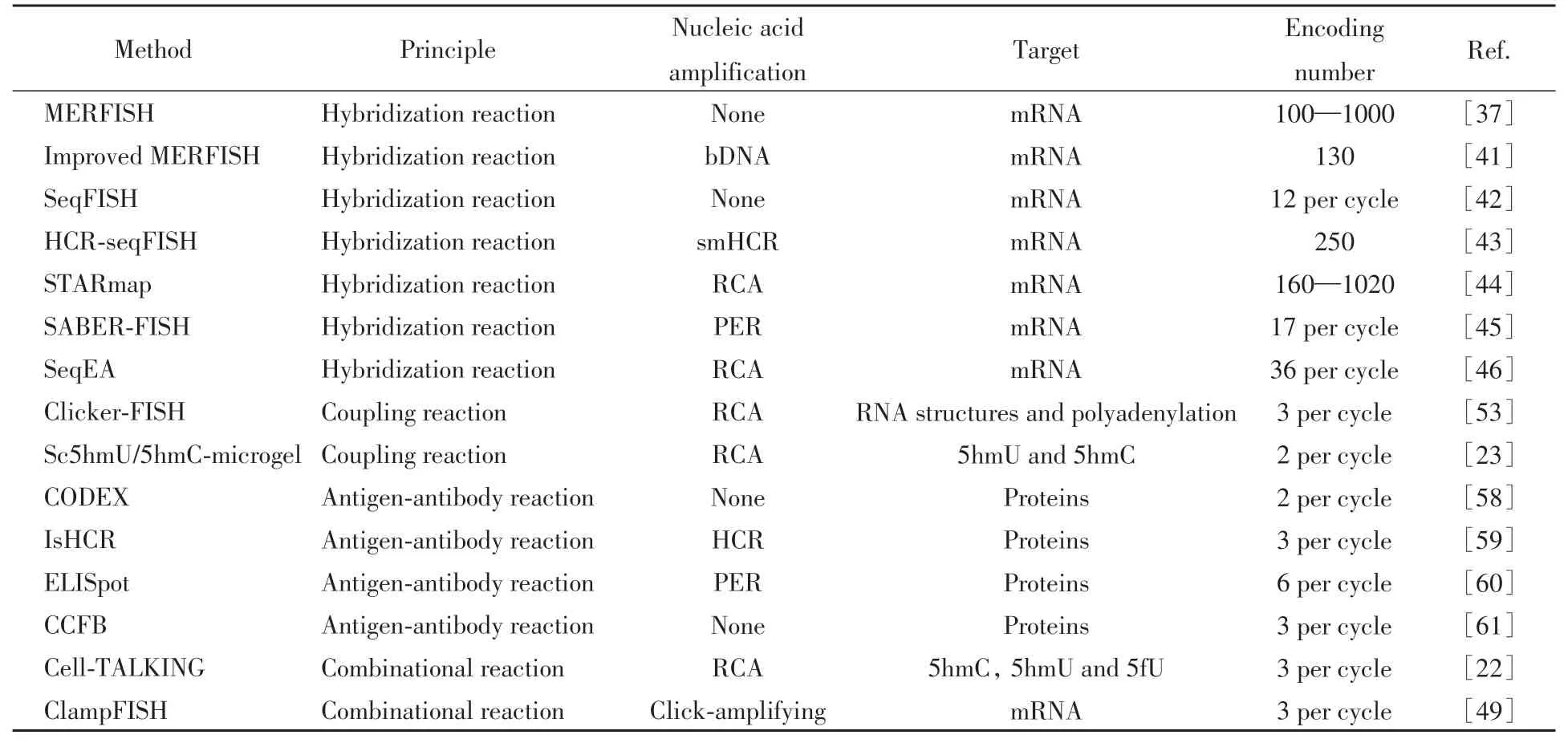
Table 1 Properties of in situ nucleic acids-encoded amplification methods
猜你喜欢
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
条码与信息系统(2021年1期)2021-12-05
装备制造技术(2020年1期)2020-12-25
商品与质量(2020年46期)2020-11-26
中国(俄文)(2020年8期)2020-11-23
条码与信息系统(2020年5期)2020-06-07
中国与非洲(法文版)(2015年4期)2015-11-09
印刷技术·数字印艺(2014年7期)2014-08-27
