
粘接层预氧化对热障涂层TGO生长与抗高温氧化性能的影响
2023-01-16 05:04:22安国升李文生李子钰
兰州理工大学学报 2022年6期
安国升, 李文生*, 冯 力, 成 波, 李子钰, 周 兰
(1. 兰州理工大学 材料科学与工程学院, 甘肃 兰州 730050; 2. 兰州理工大学 省部共建有色金属先进加工与再利用国家重点实验室, 甘肃 兰州 730050; 3. 兰州理工大学 机电工程学院, 甘肃 兰州 730050)
热障涂层(thermal barrier coating,TBC)技术的飞速发展,使其广泛应用在航空发动机与地面燃气轮机叶片、燃烧室等热端部件,通过降低工件表面温度,实现各部件的高效长寿命工作[1-2].典型的热障涂层由陶瓷层、金属粘接层与合金基体组成[3],通常采用大气等离子喷涂技术(atmospheric plasma spraying,APS)制备[4-5].但是在高温工况下,大气中的氧可通过TBC陶瓷层的喷涂缺陷,快速渗透并到达粘接层表面发生氧化反应[6],从而导致热生长氧化物(thermally grown oxide,TGO)在陶瓷层/粘接层界面处形成并持续生长[7].单位时间内,到达反应界面的氧越多,则氧化反应速率越快,生成TGO的厚度越大.当TGO达到8~12 μm的临界厚度时,将严重威胁TBC的使用寿命[8].
TGO的生长过程大致分为3个阶段:Al2O3急速生长、Al2O3稳定生长,混合氧化物的出现及其快速增厚[9].氧化初期,首先形成的Al2O3层具有较低的氧离子扩散速率,有利于抑制TGO的生长,但该阶段Al2O3层的快速增厚却缩短了氧化后期TGO层到达临界厚度的时间,即加速了TBC的失效.而在氧化后期,TGO层厚度过大,特别是混合氧化物的出现及其持续生长,将引起陶瓷层与粘接层层间热失配应力不断积累,导致TBC产生横向裂纹,甚至诱发涂层剥落[10].由此可见,TGO层的形成与生长是导致TBC失效的重要诱因之一[11].
相关研究表明,单一致密的Al2O3层是有助于减缓涂层氧化的最理想TGO结构[12].本文采用真空预氧化技术在金属粘接层表面制备了致密Al2O3层,通过 1 100 ℃高温氧化试验,研究了预氧化层在高温氧化过程中对TGO生长的影响规律.
1 试验材料与方法
将Inconel 738加工为φ25.4 mm×3 mm的基体试样,通过俄罗斯生产的爆炸喷涂设备(АДМ-4Д)制备NiCoCrAlY(Amdry 365-2,-75+45 μm)粘接层,采用国产APS设备(GP-80,九江贝斯特等离子喷涂公司)制备YSZ(Y2O3部分稳定ZrO2,METCO 204B-NS,-75+38 μm)陶瓷层;随后,将一部分喷涂态试样放置于管式炉(GSL-1500X-50,合肥科晶材料技术有限公司)进行真空预氧化处理(1 050 ℃,保温2 h,真空度为1.0×10-3Pa,升温速率为 5 ℃/min).最终获得经过预氧化P-TBC(Preoxidation-TBC)与喷涂态A-TBC(As sprayed-TBC)两组试样.涂层喷涂制备参数见表1、2.
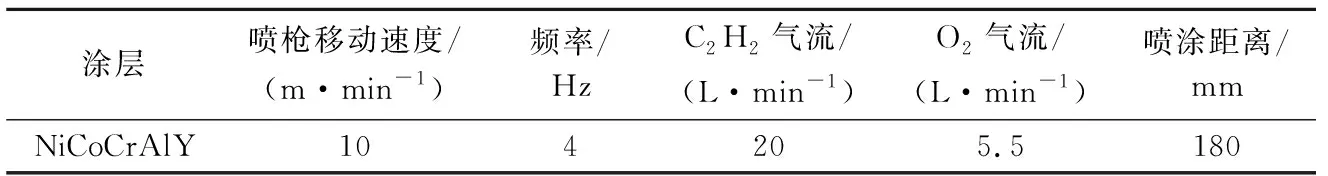
表1 爆炸喷涂工艺参数[13]Tab.1 Parameters of detonation gun spraying

表2 大气等离子喷涂工艺参数[13]Tab.2 Parameters of atmospheric plasma spraying
高温氧化试验在恒温1 100 ℃的马弗炉中进行,设置其升温与降温时间均为10 ℃/min,P-TBC与A-TBC试样氧化时间均为10、25、50、100 h.每个氧化时间段,通过电子天平(精度0.1 mg,Mettler Toledo)计算3个平行样品的平均值,获得氧化增重数据.
采用带有X射线能谱仪(EDS)的扫描电子显微镜(SEM,Quanta FEG 450)获得涂层微观形貌与元素分布;不同氧化时间的TGO生长厚度平均值,由Image-Pro Plus 6.0软件统计并计算同一时间10张SEM截面图而获得.
2 分析与讨论
2.1 氧化前微观形貌
图1为A-TBC与P-TBC氧化前截面微观形貌.可以看出,两组试样均由厚度为(250±20) μm的YSZ层、(110±20) μm的NiCoCrAlY层以及金属基体组成,分别如图1a、b所示.同时,两组陶瓷层均呈现APS典型的层状结构与较多孔隙,A-TBC与P-TBC的孔隙率分别达到13.2%与11.8%.这是因为在采用APS技术制备陶瓷层过程中,由等离子体极速加热(200~600 m/s)[14]的陶瓷粉末存在部分半熔融粒子,当这些粒子与已沉积涂层接触凝固收缩时,由于层与层之间粒子的不完全堆叠形成可以留存部分气体(包括大气与喷涂气体)的狭小空间,而这些空间被再次喷涂的熔融粒子撞击时,留存的气体会在涂层中不完全扩散,导致涂层中形成大小不一的孔隙[15];而大部分完全熔化的陶瓷粒子则层层堆叠,形成最终的层状结构.最终,以孔隙为代表的喷涂缺陷为氧在陶瓷层的扩散提供了便捷的通道,对热障涂层整体的抗高温氧化性能极为不利.
同时发现,A-TBC与P-TBC的陶瓷层/粘接层界面处均为不规则波浪状.但值得注意的是,与A-TBC不同,经过真空预氧化处理的P-TBC在YSZ/NiCoCrAlY界面处呈现明显的黑色氧化物分布,如图1b所示.通过进一步放大其界面红色虚线框区域形貌发现,该氧化物为厚度约0.75 μm的致密层;经过EDS能谱分析可知,该氧化层为 Al、O元素高度重叠区域,即为Al2O3层,如图1c所示.说明通过真空预氧化处理已经在P-TBC的YSZ/NiCoCrAlY界面处形成了稳定致密的Al2O3层.此外,在P-TBC的粘接层靠近YSZ/NiCoCrAlY界面区域,呈现深灰色与浅灰色两种颜色分布,根据点a的EDS扫描结果可知,深灰色区域Al、Ni元素高度富集,而周围浅灰色区域Al含量则明显减少.结合EDS点扫描结果与已有文献研究表明[16],深灰色区域为β-NiAl,浅灰色区域为粘接层的贫Al区.粘接层出现这种形貌源于Al在真空预氧化中参与反应生成Al2O3层;同时,较多β-NiAl的存在说明真空预氧化消耗的Al较少,即生成的Al2O3层厚度较小.

图1 氧化前A-TBC与P-TBC截面微观形貌Fig.1 Cross-section morphology of A-TBC and P-TBC before oxidation
2.2 TGO层的形成与生长
图2为1 100 ℃氧化10 h时A-TBC与P-TBC的陶瓷层/粘接层界面区域微观形貌.如图2a所示,与氧化前相比,A-TBC的界面区域有明显Al2O3层形成,其厚度达到1.70 μm;此外,A-TBC粘接层内靠近TGO一侧出现大面积贫Al区域,仅保存有少数β-NiAl.说明氧化10 h的A-TBC粘接层有较多Al参与了氧化反应,造成TGO的厚度在此阶段快速增长.但是,经过真空预氧化处理的P-TBC在氧化10 h展现了不同的形貌特征,如图2b所示,尽管受到氧化反应影响,在P-TBC的陶瓷层/粘接层界面区域的Al2O3层TGO厚度也有所增长,达到1.18 μm,但与氧化前已有的Al2O3层相比,TGO厚度增幅明显小于A-TBC.而且,相比于图2a,P-TBC粘接层贫Al区域更小、β-NiAl更多,说明前10 h的P-TBC氧化速率明显低于A-TBC,即真空预氧化处理抑制了TGO在氧化初期的快速增厚.

图2 A-TBC与P-TBC经过1 100 ℃氧化10 h截面微观形貌Fig.2 Cross-section morphology of A-TBC and P-TBC after oxidized at 1 100 ℃ for 10 h
图3展示了A-TBC与P-TBC在1 100 ℃恒温氧化100 h时的陶瓷层/粘接层界面区域微观形貌.可以看出,经过长时间氧化,A-TBC的TGO形貌演变为上层灰色、下层黑色的双层结构,通过对上层灰色区域b点进行元素分析发现,该区域为Al、Cr、Co、Ni的混合氧化物层,如图3a所示的EDS扫描结果,而TGO下层依然为黑色的Al2O3层;此时,混合氧化物层与Al2O3层的厚度分别为2.09、3.90 μm,使得A-TBC的整体TGO厚度达到5.99μm.
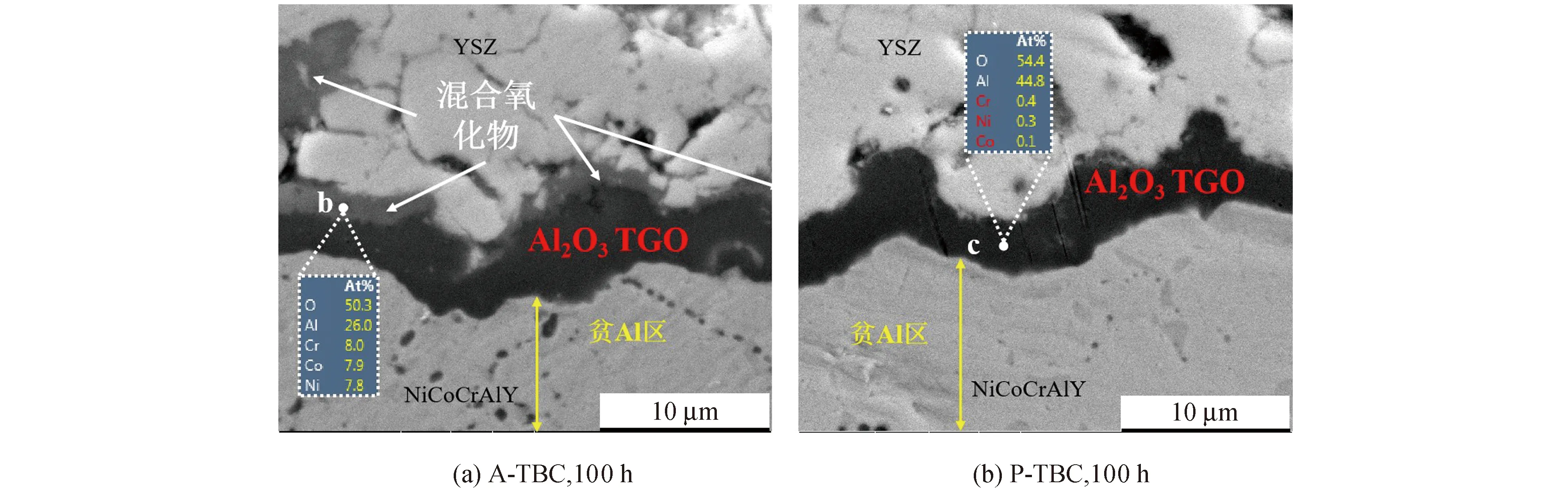
图3 A-TBC与P-TBC经过1 100 ℃氧化100 h截面微观形貌Fig.3 Cross-section morphology of A-TBC and P-TBC after oxidized at 1 100 ℃ for 100 h
但是,P-TBC在相同实验条件下,其TGO形貌依然保持单一的黑色Al2O3层,如图3b点c的EDS扫描结果.此外,P-TBC中整体TGO层厚度相比A-TBC明显较小,仅为5.01 μm.不过,由于氧化时间较长,A-TBC与P-TBC粘接层中均有大量Al元素参与反应,因此,两组试样粘接层可视范围内均呈现贫Al状态.
为了更清楚地描述两组TBC的氧化状况,绘制A-TBC与P-TBC在1 100 ℃恒温氧化0~100 h的TGO厚度变化曲线,如图4所示.可以看出,A-TBC在0~10 h内经历了Al2O3的急速生长阶段,其TGO厚度由0迅速增长至1.70 μm,如图中黑色阶段Ⅰ所示.而在A-TBC的第Ⅱ阶段,由于已形成Al2O3层对氧离子扩散的抑制作用,使得A-TBC的Al2O3生长速率明显降低,所以在10~50 h阶段TGO趋于稳定生长,其50 h的TGO厚度为3.22 μm.
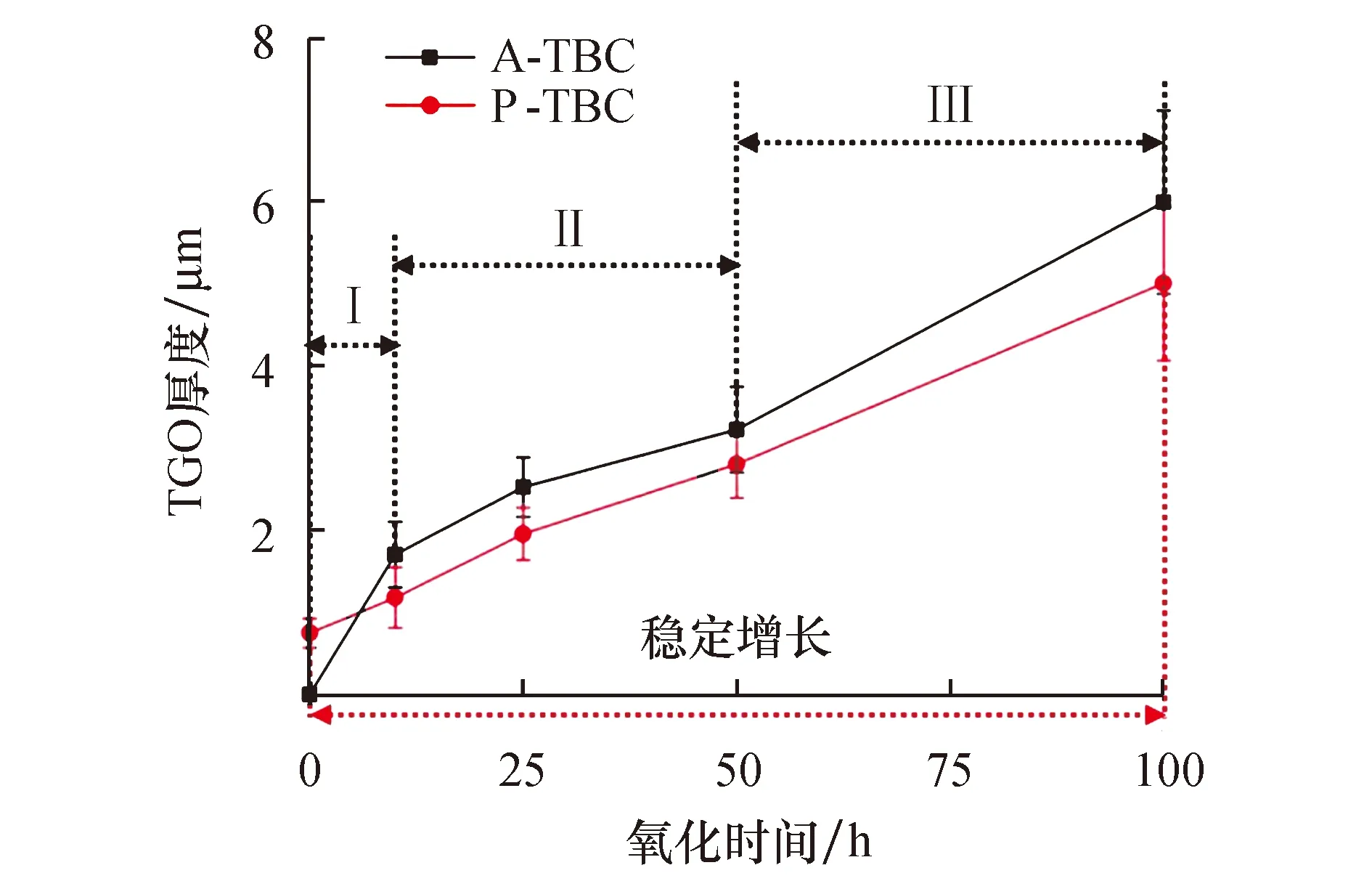
图4 TGO厚度随时间变化曲线Fig.4 TGO thickness as a function of oxidation time
但是,当氧化时间超过50 h,A-TBC的TGO生长速率又明显上升,并在Al2O3层低速生长的同时开始出现快速生长的混合氧化物,最终100 h时形成上层灰色混合氧化物、下层黑色Al2O3的双层TGO形貌;与氧化50 h相比,A-TBC在第Ⅲ阶段的Al2O3层厚度仅增长0.68 μm,而混合氧化物厚度则从无到有达到2.09 μm,说明在50~100 h的氧化过程中,A-TBC经历了混合氧化物的出现及其快速增厚阶段.
根据图4中红色曲线所示P-TBC的TGO厚度变化可以发现,基于真空预氧化处理的有益作用,P-TBC的Al2O3层经历100 h始终处于稳定生长状态.此外,在开始氧化之后,P-TBC的TGO厚度始终小于A-TBC,而且直至氧化结束,P-TBC中也未出现混合氧化物.说明通过真空预氧化处理,可以有效避免P-TBC经历Al2O3的急速生长阶段,抑制Al元素的过度消耗,减缓TGO生长速率,从而延迟混合氧化物的出现时间,达到延长TBC使用寿命的目的.
2.3 氧化动力学曲线
图5a为1 100 ℃恒温0~100 h时两组TBC的氧化增重曲线.可以发现,与TGO厚度变化几乎一致,A-TBC的氧化增重经历了0~10 h快速增重、10~50 h稳定增长、50~100 h再次快速增重的3个阶段,其100 h的氧化增重为2.44 mg·cm-2;而P-TBC由于真空预氧化处理产生0.306 6 mg·cm-2增重,导致其0 h时相比A-TBC更重,但是其余整个氧化过程中都比同期A-TBC的氧化增重更小,且始终处于稳定增长阶段,使得P-TBC最终100 h的氧化增重仅为2.30 mg·cm-2.根据氧化增重结果与Wager理论,拟合得到A-TBC与P-TBC的抛物线氧化速率Kp分别为6.08×10-2、5.18×10-2mg2·cm-4·h-1,如图5b所示.这说明经过1 100 ℃高温氧化100 h后,P-TBC的抛物线氧化速率相比A-TBC下降了14.80%,证明真空预氧化处理能够有效提高TBC抗高温氧化性能.
图5 A-TBC与P-TBC经过1 100 ℃恒温氧化100 h的动力学曲线Fig.5 Isothermal oxidation kinetics curves of A-TBC and P-TBC after oxidized at 1 100 ℃ for 100 h
3 P-TBC的抗高温氧化机理
为进一步揭示P-TBC在整个试验过程中表现出更小TGO生长厚度与更低抛物线氧化速率的原因,绘制两组TBC高温氧化过程中TGO的形成与生长机理示意图,如图6所示.由图6可知,通过真空预氧化处理,P-TBC在高温氧化实验之前已经有Al2O3层在YSZ/NiCoCrAlY界面形成,如图6中0 h情况所示.这不仅因为在NiCoCrAlY粘接层各元素当中,Al与O的亲和势最高,达到1 675.7 kJ/mol[17],而且Al2O3的生成吉布斯自由能为负值(-850.5 kJ/mol,1 273 K)[18],为氧化反应的自发形成提供了热力学条件.其反应方程为[19]:

图6 A-TBC与P-TBC高温氧化过程TGO形成与生长机理示意图Fig.6 Schematic illustration of the formation and growth mechanisms of TGO during high-temperature oxidation of A-TBC and P-TBC

(1)
此外,因为氧在涂层中的扩散通量与涂层两端氧分压差异成正比[20],所以通过真空预氧化处理形成的Al2O3层厚度非常小.尽管如此,由于相比于TGO中其他氧化物,氧在Al2O3中的扩散系数极小,见表3.因此,预氧化Al2O3层依然对氧的扩散产生了显著的抑制作用,这导致当1 100 ℃高温氧化10h时,A-TBC与P-TBC形成了厚度明显不同的氧化层.
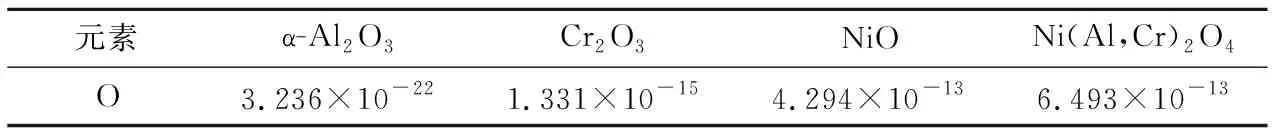
表3 氧在TGO中的扩散系数(cm2/s,1 100 ℃)[17]Tab.3 Diffusion coefficient of oxygen in TGO
当1 100 ℃高温氧化进行至25~50 h时,A-TBC的TGO生长速率虽然比前10 h有所下降,但是长时间的氧化反应不仅导致Al2O3层持续增厚,而且造成A-TBC的粘接层中Al元素大量消耗,而此时,P-TBC在前10 h较低氧扩散速率的有益作用下,其Al2O3层厚度依然较A-TBC的更小.当氧化时间超过50 h,Al在A-TBC粘接层中的过量消耗导致其他金属元素(Ni,Co与Cr)开始穿过Al2O3层参与氧化反应,且这些元素在Al2O3层的扩散速率远高于氧,因此混合氧化物形成于Al2O3层之上靠近陶瓷层的位置,其反应方程为[21]:

(2)
(3)
(4)
随后,氧化物之间继续发生反应,形成更具破坏性的尖晶石氧化物,其反应方程为[7,9,21,22]:
(5)
(6)
(7)
(8)
一旦混合氧化物形成,氧在其中的扩散速率相比Al2O3层将极速提升,见表3,这为混合氧化物的迅速增厚提供了便利条件.同时,混合氧化物的形成过程本身伴随着体积膨胀,导致TGO中存在较大的生长应力,见表4.所以,当氧化进行到100 h时,A-TBC形成的双层结构TGO将直接缩短TBC的使用寿命.而得益于前期较少的Al元素消耗,截止到高温氧化进行到100 h,P-TBC的粘接层中Al浓度依然较高,可以继续通过氧的低速扩散实现Al2O3层的稳定生长,因此,P-TBC的TGO层仍然保持单层Al2O3结构.这说明真空预氧化处理可以通过抑制TGO形成初期的急速生长,降低Al的消耗速率,使得P-TBC长时间保持有益的单一Al2O3层,从而提高TBC抗高温氧化性能.

表4 TGO中不同氧化物的生长应力 [18]Tab.4 Growth stress of different oxides in TGO
4 结论
1) 在1 100 ℃高温氧化实验过程中,A-TBC与P-TBC 的TGO厚度与单位面积氧化增重均随氧化时间的延长而增长.但是,氧化实验进行至100 h时,A-TBC的TGO厚度为5.99 μm,并呈现为混合氧化物在上、Al2O3在下的双层结构,而P-TBC的TGO厚度仅为5.01 μm,其结构则仍然保持单一的Al2O3层.通过拟合计算可知,P-TBC的抛物线氧化速率相比A-TBC下降14.80%.
2) A-TBC的TGO在整个实验过程中经历了Al2O3的急速生长、稳定生长与混合氧化物快速生长3个阶段.但是,P-TBC的预氧化层凭借较低的氧扩散系数显著降低了0~10 h时TGO的生长速率,并使得P-TBC的TGO在整个实验过程始终处于Al2O3的稳定生长阶段,从而有效避免了粘接层中Al的过度消耗,最终有效提高了P-TBC的抗高温氧化性能.
猜你喜欢
江苏安全生产(2023年11期)2023-12-14 12:05:26
真空与低温(2022年6期)2023-01-06 07:33:20
陶瓷学报(2020年6期)2021-01-26 00:37:56
中学生数理化·中考版(2018年11期)2019-01-31 06:18:06
教学考试(高考化学)(2018年5期)2018-12-06 07:21:56
材料科学与工程学报(2016年4期)2017-01-15 13:35:34
橡胶工业(2015年6期)2015-07-29 09:20:34
中国光学(2015年1期)2015-06-06 18:30:20
中国卫生(2014年11期)2014-11-12 13:11:20
郑州大学学报(工学版)(2014年6期)2014-03-01 04:21:27
