
基于网络拓扑和多种生物信息融合的关键蛋白质识别算法
2023-01-16 05:03:40卢鹏丽陈云天
兰州理工大学学报 2022年6期
卢鹏丽, 陈云天
(兰州理工大学 计算机与通信学院, 甘肃 兰州 730050)
关键蛋白质是指通过基因剔除式突变将其移除后,造成生物体相关功能缺失,如衰竭、异常生长或丧失生命力的蛋白质[1].关键蛋白质的准确鉴定和分析,对生物学进化、疾病的预防和新药物的研发有着重要意义[2].在生物医学中,依靠生物实验识别关键蛋白质的方法有单基因敲除[3]、RNA干扰和条件基因敲除[4].通过生物学实验预测关键蛋白质虽然成果有效且准确性高,但是需要耗费大量的成本和资源.
由于“中心性-致死性”[5]的提出,许多中心性方法[6]被用于关键蛋白质的识别.目前基于蛋白质相互作用网络(PPI)拓扑特性提出的中心性算法包括:度中心性(DC)通过计算邻居节点的个数判断节点的重要性[7];介数中心性(BC)通过经过该节点最短路径的数目判断其重要性,如果BC值较高,说明该节点对整个网络信息传输产生的影响较大,节点处于关键枢纽位置[8];子图中心性(SC)通过网络局部特征分析节点重要性[9];特征向量中心性(EC)表明节点重要性不仅依赖于邻居节点的数量,而且与每一个邻居的重要性也有关[10];局部平均度(LAC)考虑了节点在诱导子图中的特性[11];网络中心性(NC)基于边聚类系数,通过复杂的连边关系判断节点重要性[12].
仅依赖网络节点的拓扑特性不足以准确地识别关键蛋白质,不少研究者利用生物信息来提高识别关键蛋白质的准确率[13].目前用于识别关键蛋白质的生物信息包括蛋白质的基因本体论[14]、亚细胞定位[15]、基因表达序列[16]和蛋白质复合物信息[17]等.随后提出了一系列结合生物信息的识别方法.如GEG方法同时利用了语义相似性和基因表达序列来丰富蛋白质的信息[18].联合复合物中心性(UC)考虑了蛋白质出现在不同蛋白质复合物中的频率,并结合了边聚类系数提升识别的准确率[17].局部相互作用密度中心性(LIDC)基于网络拓扑和复合物信息[19].PEC和WDC都基于边聚类系数和基因表达序列[20-21]来识别关键蛋白质.
本文基于网络拓扑结构和多源生物信息提出了关键蛋白质识别算法.首先,通过考虑节点自身特性以及节点与邻居节点间存在的三角形个数来衡量网络拓扑结构对节点的影响,当蛋白质网络的拓扑结构不能区分蛋白质的关键性时,考虑蛋白质的生物特性.本文根据不同亚细胞中的蛋白质参与不同生命活动这一特性[22],计算亚细胞定位分值,利用亚细胞定位分值为每个蛋白质赋予权值,提出了SNC(subcell_nodecentrality)方法.经过分析,发现网络中一些结构为星型的蛋白质的SNC值为零,为更准确地区分蛋白质节点的关键性,将复合物信息与亚细胞定位信息相结合提出了SIDC(subcell_indegree centrality)方法.最后,通过赋予不同比重的SNC和SIDC,提出了CTB(combinationtopology_bioinformation)算法来实现网络拓扑与多源信息融合去识别关键蛋白质.比重的赋予是通过分析网络的局部拓扑结构,按照邻居间是否存在连边,将节点划分为三角形类Ttype(邻居间存在连边)和星型类Stype(邻居间不存在连边)两种,最终计算Ttype类型的节点在网络中的占比来赋予比重.文中所对比的九种算法中,前六种仅依赖网络拓扑结构,忽略了蛋白质自身携带的生物属性.算法PEC和WDC仅使用一种生物信息,结果不够精确.相比于问题中涉及到的对比算法,本文提出的CTB算法有两方面优势:一方面,从网络拓扑出发,解决了部分蛋白质由于自身结构特殊导致不可对其进行关键性判断的问题;另一方面,通过融合生物信息,解决了当蛋白质拥有相同拓扑结构而不能明确区分其关键性的问题.为了评估CTB算法的性能,在YDIP、YMIPS和Krogan三种蛋白质网络数据集上进行实验,通过与已有的九种算法(BC、DC、SC、LAC、EC、UC、NC、PEC和WDC)进行对比,实验结果表明CTB算法能够更有效地识别关键蛋白质.
1 相关工作
1.1 预备知识
蛋白质相互作用网络可以看作是一个简单图G(V,E),其中V(G)={v1,v2,…,vn}表示顶点集,E(G)={e1,e2,…,em}表示边集,图G的顶点数n=|V(G)|,边数e=|E(G)|.Z(u,v)表示由u、v及其公共邻居节点形成的三角形的数量.蛋白质对应图中的顶点,蛋白质之间的相互作用对应图中的边.dv表示节点v的度,记作dv=|Nv|,Nv表示节点v的邻居集合.
1.2 已有方法
(1) 度中心性DC(degree centrality)[7]:
(2) 介数中心性BC(betweenness centrality)[8]:
其中:σst是指从节点s出发到达终止节点t的最短路径数目;σst(v)表示从节点s出发到达终止节点t,且通过节点v的最短路径数目.
(3) 特征向量中心性EC(eigenvector centrality)[10]:
EC(v)=αmax(v)
其中:αmax是对应于网络邻接矩阵的最大特征值λmax的特征向量;αmax(v)是αmax的第v个分量.
(4) 网络中心性NC(network centrality)[12]:
(5) 联合复合物中心性UC(united complex centrality )[17]:
其中:fu表示节点u在不同复合物中出现的次数;fM表示蛋白质节点在复合物中出现的最大次数.
2 本文提出的算法
2.1 SNC算法
通过综合衡量节点自身在网络中的拓扑特性以及邻居节点之间的复杂连边关系,提出了点-边中心性方法NAEC.该方法考虑了节点间紧密程度,且利用节点与其邻居节点间构成的三角形个数来量化节点间边的重要性,具有更全面的拓扑特征,公式如下:
其中:E(v)表示节点v的邻居节点间实际具有的边数.一个节点的NAEC(v)值越大,则表示节点v与邻居节点间的连接更紧密,进一步表明节点越倾向于形成高度连接的簇,更有可能成为关键的蛋白质.
亚细胞定位信息是指定位生物大分子如蛋白质在细胞内的存在的具体位置.生物体中存在十一种亚细胞,不同亚细胞中的蛋白质功能不同,因而蛋白质的重要程度也不同.出现在越多亚细胞中的蛋白质表明它参与越多的生物进程,成为关键蛋白质的概率更大.亚细胞定位分值(subcellular score,SC)作为一种衡量蛋白质关键性的测度指标,由下式计算得出:
其中:SC(i)表示蛋白质网络中的所有节点出现在第i个亚细胞中的数目,i∈{1,2,3,…,11};SCtotal表示整个网络中的所有节点出现在11种亚细胞中的总数.
若两个蛋白质网络拓扑结构相同,则其对应NAEC值相同,因此不能区分其重要性.文中提出SNC采用网络拓扑特性与亚细胞定位信息相融合的方式来解决这一问题.若两个蛋白质网络结构相同,可通过SC得到对应的蛋白质亚细胞定位分值,从而提升识别关键蛋白质的准确率,公式如下:
SNC(v)=SC(v)*NAEC(v)
2.2 SIDC算法
通过对网络结构进行分析,发现网络中一些结构为星型的蛋白质的SNC值为零,为更准确地区分蛋白质节点的关键性,将复合物信息与亚细胞定位信息相结合提出了SIDC方法.该方法不仅考虑了蛋白质在复合物中的局部度中心性以及出现频率,也考虑了蛋白质在亚细胞定位信息中的全局属性.由于复合物中的蛋白质是可以在相同时间和空间上相互协作特定功能的蛋白质,本文结合蛋白质在复合物中子集中的特性,更加全面地挖掘蛋白质的生物特性.公式如下:

其中:ComplexSet(v)表示所有包含蛋白质v的复合物子集;In-Degree(v)i表示蛋白质v在第i个复合物中的度值,定义为
In-Degree(v)i=DC(v)i
DC(v)i是蛋白质v在第i个复合物中的度值.利用复合物构成的社团结构中节点较网络全局节点更加稠密这一特性,可以提升度中心性的性能.
2.3 CTB算法
为了实现网络拓扑与多源生物信息的融合,将不同比重的SNC与SIDC方法相结合提出了关键蛋白质识别算法CTB.为合理分配两种方法的比重,基于节点的局部拓扑结构,按照邻居节点间是否存在连边将蛋白质节点分为三角形类Ttype和星型类Stype两类.通过计算两种类型蛋白质在网络中的占比来获得参数,若网络中Ttype类蛋白质数量多,则SNC算法对应的参数值较大,即SNC算法更能影响蛋白质的关键性,反之SIDC更能影响蛋白质的关键性.CTB算法表示如下:
其中:SNCmax表示SNC(v)的最大值;SIDCmax表示SIDC(v)的最大值.在YDIP、YMIPS和Krogan这三种不同的网络中,三角形类Ttype和星型类Stype的蛋白质占比不同,分别对应不同的β值.β的值由如下公式得到:
其中:|Ttype|表示网络中三角形类Ttype蛋白质的数量;n表示网络中的蛋白质总数.
3 实验结果与分析
3.1 实验数据来源
(1) PPI网络数据:选择相对完整可靠的蛋白质相互作用网络作为实验数据,包括YDIP[24]、YMIPS[25]和Krogan[26].通过去除重边和自相互作用后,得到的网络数据如表1所列.
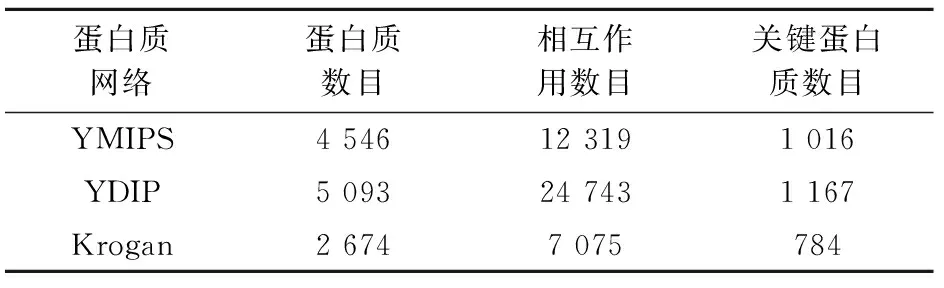
表1 蛋白质网络YDIP、YMIPS和KroganTab.1 The PPI network YDIP、YMIPS、Krogan
(2) 标准关键蛋白质数据:来自数据集MIPS[25]、SGD[27]、DEG[28]和SGDP[1].
(3) 蛋白质复合物信息:选择来自CM270[25]、CM425[29]、CYC408[30]和CYC428[31]的745个蛋白质复合物子集.
(4) 亚细胞定位信息:下载自COMPARTMENTS数据库,包含11种亚细胞定位信息[32].
3.2 评估对比分析
3.2.1使用六种评估方法对比分析
为了评估算法的性能,首先通过CTB算法判断蛋白质的重要性并降序排列,选取前20%的蛋白质作为候选关键蛋白质,剩余的80%作为候选非关键蛋白质.通过对比标准关键蛋白质数据,可以得到候选关键蛋白质中被正确识别为关键蛋白质的数目.使用了六种评估方法,包括准确率(ACC)、F-度量(F-measure)、阳性预测值(positive predictive value,PPV)、阴性预测值(negative predictive value,NPV)、敏感度(sensitivity,SN)和特异性(specificity,SP),计算方式如下:
其中:真正例(TP,true positive)指候选关键蛋白质中被正确识别为关键蛋白质的数目;假正例(FP,false positive)指候选非关键蛋白质被错误识别为关键蛋白质的数目;假反例(FN,false negative)指候选关键蛋白质被错误识别为非关键蛋白质;真反例(TN,true negative)指候选非关键蛋白质被正确识别为非关键蛋白质的数目.
以上六种统计指标可以综合评估CTB的性能,若算法对应的指标值越大,说明算法性能越优.通过对比BC、DC、SC、LAC、EC、UC、NC、PEC和WDC九种方法评估算法性能,实验结果见表2,结果表明CTB算法的六项评估指标均优于其他方法.

表2 六种统计指标对比结果Tab.2 Comparison results of six statistical indicators
3.2.2关键蛋白质识别数目比较
在YDIP、YMIPS和Krogan三种蛋白质网络上实现了BC、DC、SC、LAC、EC、UC、NC、PEC、WDC和CTB算法,并将网络中的蛋白质按照其重要性排序.选取前100~600的蛋白质作为候选集,再对比标准关键蛋白质,得出候选关键蛋白质中真正的关键蛋白质数量,实验结果如图1~3所示,图中横坐标1~10分别代表DC、BC、EC、SC、LAC、NC、UC、PEC、WDC和CTB算法.

图1 CTB与已有算法在YMIPS网络中的对比Fig.1 Comparison of CTB and existing algorithms in YMIPS networks
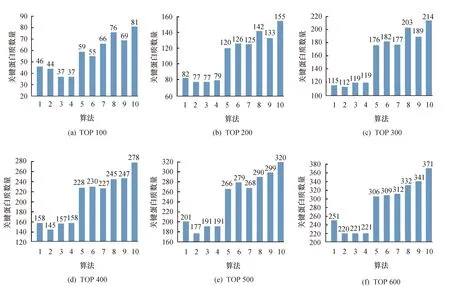
图2 CTB与已有算法在YDIP网络中的对比Fig.2 Comparison of CTB and existing algorithms in YDIP networks
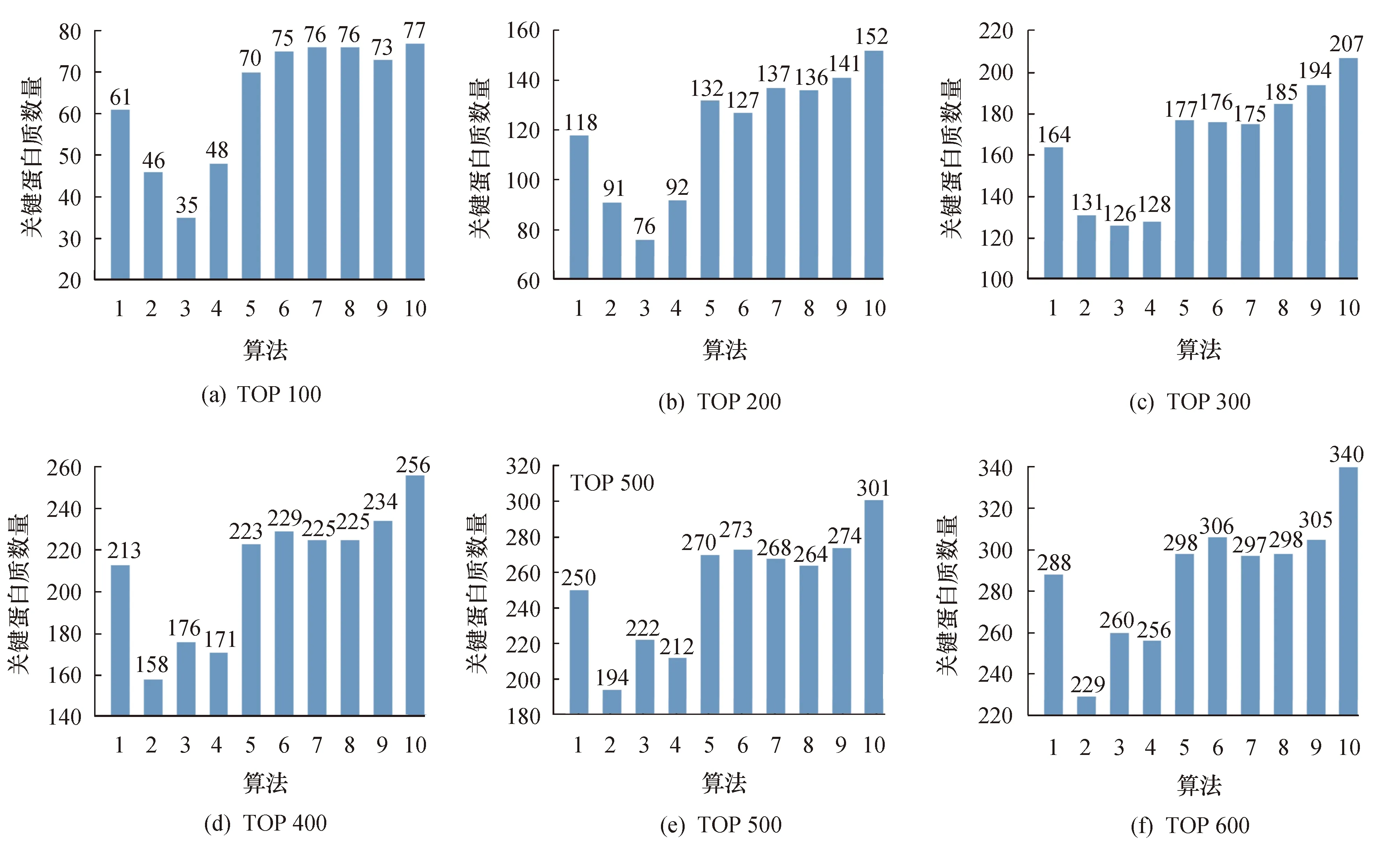
图3 CTB与已有算法在Krogan网络中的对比Fig.3 Comparison of CTB and existing algorithms in Krogan networks
CTB算法识别出真正的关键蛋白质数量明显多于其他算法,尤其在YMIPS网络中,CTB在前600个候选蛋白质中,正确识别出337个关键蛋白质,较PEC算法多出118个.因而,CTB算法具备更高效准确的性能.
3.2.3参数β和亚细胞定位信息对算法的影响
本文提出的算法CTB受到参数β和亚细胞定位信息的影响,本节分析这两种因素对算法CTB性能的影响.将不含参数β的CTB算法记作CTB-1,将不考虑亚细胞定位信息的CTB算法记作CTB-2,将不含参数、亚细胞定位信息的CTB算法记作CTB-3.在Krogan、YDIP和YMIPS网络中,选取前100~600候选关键蛋白质,对比CTB、CTB-1、CTB-2、CTB-3的性能,实验结果如图4所示.
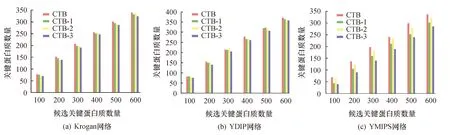
图4 参数及亚细胞定位信息的影响Fig.4 The influence of parameters and subcellular localization information
由实验结果可以看出,融合亚细胞定位信息且考虑到参数影响后,算法CTB的性能较其他三种情况有明显提升.
3.2.4P-R曲线评估
查准率(Precision)-查全率(Recall)曲线(P-R曲线)的x轴代表查全率,y轴代表查准率.该评估方法中,曲线越高,表明算法性能越优.本文在三种网络上利用精准召回曲线比较算法的性能,实验结果如图5所示.在三种网络中,由CTB算法计算得到的P-R曲线均高于其他九种方法,表明CTB算法的性能优于其他算法.查全率和查准率计算方式如下:

图5 P-R曲线在三种网络中的对比Fig.5 Comparison of P-R curves in three networks
4 结论
关键蛋白质的研究能够促进生物医学的发展.为更加准确地识别关键蛋白质,首先考虑节点自身及其复杂的连边关系来表征网络的拓扑特性,并结合亚细胞定位信息,提出了SNC方法.为更好地区分蛋白质节点的关键性,将复合物信息与亚细胞定位信息相结合提出了SIDC方法.最后,通过不同比重的SNC和SIDC方法来实现网络拓扑与多源信息融合,提出了CTB算法来识别关键蛋白质.本文选用YMIPS、YDIP和Krogan三种蛋白质网络数据,运用多种评估方法与已有算法BC、DC、SC、LAC、EC、UC、NC、PEC和WDC进行对比,实验结果表明,CTB算法识别蛋白质的性能高于已有的九种识别方法,能够有效提高识别关键蛋白质的准确率.
猜你喜欢
网络安全与数据管理(2022年2期)2022-05-23 13:25:46
中学生数理化(高中版.高考理化)(2021年6期)2021-07-28 06:21:04
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
电子制作(2018年23期)2018-12-26 01:01:16
中成药(2018年7期)2018-08-04 06:04:18
中成药(2018年3期)2018-05-07 13:34:18
汽车维修技师(2017年10期)2017-03-17 02:25:01
电测与仪表(2016年5期)2016-04-22 01:13:46
NBA特刊(2014年7期)2014-04-29 00:44:03
中国商人(2013年1期)2013-12-04 08:52:52
