
长期饮用白酒中度扰动肠道微生物多样性演替研究
2023-01-07 14:34张传旭宫田润韩靖昱胡大伟
中国酿造 2022年12期
张传旭,张 艺,宫田润,韩靖昱,胡大伟,董 琛,*
(1.山东体育学院 运动营养与智慧配餐实验室,山东 济南 250102;2.山东体育学院 健康服务与管理教研室,山东 济南 250102;3.北京航空航天大学 生物与医学工程学院,北京 100191)
长期高频度大量饮酒会引起人体肠道微生物的紊乱,并增加患病风险[1-2]。肠道作为人体最重要的菌库之一,其微生物多样性是评价健康与否的重要指标。酒精对细菌直接的毒副作用、改变肠道的应激反应和提供乙醇作为细菌代谢底物等方式易导致肠道微生物菌群失调,影响营养吸收效率[3-4]。与体外环境相比,肠道中可供微生物生长的基质无论从种类还是数量上来看,都是有限的。肠道内分泌消化液,其消化是食物消化吸收最重要的环节。与体外环境相比,肠道中可供微生物生长的基质无论从种类还是数量上来看,都是有限的。根据生态学中著名的竞争排斥原理,同一营养级上生物的种类数目不能多于可利用底物的数目,否则会产生剧烈的竞争排斥,直到不多于底物数目的优势种类经生态位分化后,可以最终共存不可能形成和维持生物多样性[5]。在已有研究基础上,尽管在酒精刺激下,肠道中发现了微生物多样性演替变化的现象,但是其形成和维持背后的演替动力学机制目前仍然没有得到合理的解释[6]。本研究选择了人体肠道最常见的粪杆菌属(Faecalibacterium)、拟杆菌属(Bacteroides)、罗氏菌属(Roseburia)、双歧杆菌属(Bifidobacterium)和布劳特氏菌属(Blautia)5个菌属,比较它们在有、无长期饮酒干预下的相对丰度等数据,通过系统动力学的方法建立了随机微分方程组描述微生物演替的动力学机制,利用大量的Monte Carlo仿真实验证实了结果和模型的有效性,为提高饮酒健康建立坚实的营养学、生理学基础。
1 对象与方法
1.1 对象
本研究的肠道微生物样本来自山东省的40名志愿者,长期饮酒组和空白对照组各20名,年龄在30~40岁之间。长期饮酒判断标准:饮酒史超过3年,平均每周超过3次,每次饮用酒精量>30 g,饮用种类为白酒,白酒品牌不限。空白对照组在监测期内禁止饮酒。受试者均为汉族,身体强壮,无长期代谢性疾病、无长期吸烟史、无药物依赖、无毒品成瘾史。实验前,受试者的各项体检指标基本在正常范围内,心理、情绪状态良好,无行为障碍,且近1年内没有进行过任何其他医学治疗。
微生物样本来自长期饮酒组和空白对照组的新鲜粪便样本,每隔8~12 d取样一次。每人每次收集2份相同的粪便,一份用于宏基因组分析,另一份用于实验室成分分析。针对测试结果,对肠道中常见的5种菌属[7-9],即粪杆菌属(Faecalibacterium)、拟杆菌属(Bacteroides)、罗氏菌属(Roseburia)、双歧杆菌属(Bifidobacterium)和布劳特氏菌属(Blautia)进行了4个月的监控。本研究样本来源于人体粪便,通过了山东体育学院生物与医学伦理委员会的审批,并征得了受试志愿者同意。
1.2 微生物的相对丰度和多样性分析
使用苯酚/三氯甲烷方法进行DNA提取,在BGI-SEQ500平台上进行宏基因组学测序(插入片段350 bp,读取长度100 bp)。宏基因组装和预测、样本的物种注释由上海派森诺生物科技有限公司协助完成。使用DIAMOND软件将Unigenes与京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)数据库功能数据库进行比对(Blastp,e value≤1e-5),共有638 981(63.72%)个基因能够比对上KEGG数据库,其中,有301 512(30.09%)个基因能够比对上数据库中的5 111个KEGG ortholog group(KO)。KO的相对丰度是通过使用每个样品映射上的Reads将每个KO的基因相对丰度相加而确定的[10-11]。为了得到更多的采样数据用于建模、仿真、模型参数辨识,采用B-样条插值的方法,生成关于“时间-数据”之间精确的3阶B-样条函数,保证内插预测是高度精确的。
1.3 数学建模与数值仿真
本研究基于系统动力学原理和实验数据,建立肠道微生物群落演替的动力学模型,每一种微生物表示为一个特征参数向量,因此通过Monte Carlo实验法,可以生成大量的微生物物种[12]。在MATLAB/Simulink平台上建立肠道中微生物群落演替的计算机仿真模型,服从不同正态分布的随机参数使微生物种群的随机波动具有不同的功率谱密度,分别模拟长期饮酒组和空白对照组两种环境下微生物群落演替过程中受到的不同程度扰动[13]。
根据经典的种群Lotka-Volterra竞争方程:

从式(0)中可以看出,种内竞争强度取决于xixi,而种间竞争强度取决于xixj,而微生物种群波动随机过程的自相关函数和互相关函数的定义分别为:RXX(t1,t2)=E[xi(t1)xi(t2)]和RXY(t1,t2)=E[xi(t1)xj(t2)],因此当种群变化随机过程的自相关函数RXX与种群之间的互相关函数RXY较大时,意味着种内和种间竞争强度xixi与xixj较大,这时会有大量的物种被竞争淘汰掉,反之若种群变化随机过程的自相关函数RXX与种群之间的互相关函数RXY较小时,会有大量的物种免于竞争淘汰而共存,从而形成并维持生物多样性。因此本研究将重点分析在有、无长期饮酒时,这5种菌属的自相关和互相关函数,以及其中隐含的影响微生物群落演替的动力学机制[14]。
2 结果与分析
2.1 微生物丰度的动态特征
经过周期性的采样,肠道最常见的5个菌属分别在长期饮酒组和空白对照组的肠道内相对丰度变化不同,同时计算两个场景微生物群落的Simpson多样性指数结果见图1。基于5种微生物菌属相对丰度随机过程,得到了它们之间分别在长期饮酒组和空白对照组相对丰度的自相关函数结果见图2。
由图1可知,由于受到环境扰动,这5个菌属的相对丰度都是具有不同统计特征的随机过程。空白对照组的菌属相对丰度波动性较大,其Simpson多样性指数(S)定义为:

图1 5种肠道菌属在长期饮酒组(a)和空白对照组(b)的相对丰度及群落的Simpson指数(c)Fig.1 Relative abundance of 5 gut microbial in long-term alcohol consumption group (a) and control group (b) and their Simpson indexes (c)

式中:Pi表示第i个物种的数量与群落总个体数量之比;n表示群落中物种的数目,个。
S越小,生物多样性越高,反之则越低。长期饮酒组人群中的肠道菌群Simpson多样性指数高,物种多样性则小。
由图2可知,从统计学角度发现长期饮酒组(见图2a)的受试者肠道内5种微生物菌属相对丰度随机过程的自相关函数都比较小,说明长期饮酒组人体肠道内微生物群落中各菌群的数量基本处于平台期,群落结构保持稳定。与之相反,空白对照组(见图2b)的受试者肠道内5种微生物菌属相对丰度随机过程的自相关函数都比较大,说明肠道内微生物群落中各菌群的数量未处于平台期,群落结构不稳定,更有利于形成多样性。

图2 长期饮酒组(a)及空白对照组(b)5种肠道菌属相对丰度的自相关函数Fig.2 Autocorrelation function of relative abundance of 5 gut microbial in long-term alcohol consumption group (a) and control group (b)
从生态学的角度来看,肠道中微生物之间主要的生态关系是竞争,基于生态学著名的Lotka-Volterra竞争方程和随机过程理论,各个微生物菌属的自相关函数和它们之间的互相关函数可以反应出种内和种间竞争关系的强度,两者数值越大,意味着种内和种间竞争与种群动态特征的相关性越强。在得到5种菌属自相关函数后,为研究在不同条件下肠道菌群相互间的影响和群落演替规律,基于实验数据和菌群丰度得到长期饮酒组和空白对照组相对丰度的互相关函数分别见图3。
由图3可知,长期饮酒组(见图3a)人体肠道内5种微生物菌属相对丰度随机波动之间的互相关函数都比较小,说明受试者肠道内微生物群落中各菌群之间存在较弱的相互关系和相互作用。相反,空白对照组(见图3b)的受试者肠道内5种微生物菌属相对丰度随机过程之间的互相关函数都比较大,说明肠道内微生物群落中各菌群之间存在较大的相互关系和相互作用。
空白对照组各个微生物菌属的自/互相关函数比较大、整个微生物群落的Simpson指数都比较小(图1和图3a),说明微生物群落内种内和种间竞争关系强,微生物多样性程度较高。从图1和图3b中可以看出,长期饮酒组的情况恰恰相反,这意味着微生物种群的动态特征基本上与它们的种内和种间竞争无关,微生物多样性程度较小。

图3 长期饮酒组(a)及空白对照组(b)5种肠道菌属之间相对丰度的互相关函数Fig.3 Cross-correlation function of relative abundance of 5 gut microbial in long-term alcohol consumption group (a) and control group (b)
2.2 肠道中微生物群落演替过程的系统动力学解释
2.2.1 微生物种群生长的速率方程
假设肠道中有M种不同类型的微生物,有m种不同类型的底物可供微生物群落利用,且满足M>m,模拟底物种类缺乏引起将导致竞争排斥的情况。
因此模型的状态变量是第i种微生物种群的数量(xi)和由微生物群落分解大分子有机物所产生的第k种底物的浓度(Sk),基于微生物学中经典的Monod种群生长方程[15],因此第i种微生物种群增长的速率(vbi)可以表示为:

式中:μi表示第i种微生物种群的比生长速率;ε1(t)是服从正态分布的随机数表示环境扰动对μi的影响;Kik表示第i种微生物种群生长对第k种底物浓度的半饱和常数。
2.2.2 微生物种内和种间竞争的速率方程
根据经典的Lotka-Volterra竞争模型,第i种微生物种内竞争的速率方程(vai)可以表示为:

式中:αi表示第i种微生物的种内竞争系数;ε2(t)是服从正态分布的随机数表示环境扰动对αi的影响。
类似地,第i种微生物和第j种微生物之间的种间竞争的速率方程(vei)可以表示为:

式中:βij表示第j种微生物对第i种微生物种群生长的种间竞争系数;ε3(t)是服从正态分布的随机数表示环境扰动对βij的影响。
2.2.3 微生物自然代谢速率方程
第i种微生物的自然代谢速率(vdi)可以表示为:

式中:di第i种微生物的自然代谢系数。
2.2.4 基质的生长与消耗速率
第i种微生物分解大分子有机物,产生第k种底物的速率(vpki)可以表示为[16]:
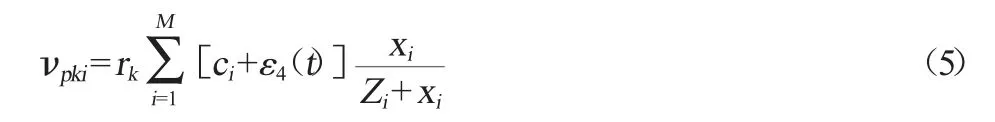
式中:ci表示第i种微生物的内禀分解制造底物的能力;Zi表示底物生产速率对自身种群数量的半饱和常数;ε4(t)是服从正态分布的随机数表示环境扰动对ci的影响;rk表示肠道的微生物群落制造的所有底物中第k种底物所占的比例。
类似地,第i种微生物在种群生长过程中,消耗的第k种底物的速率(vcki)可以表示为:

式中:γi表示第i种微生物在种群生长过程中,对底物的消耗系数;ε5(t)是服从正态分布的随机数表示环境扰动对γi的影响;hk表示微生物群落消耗的所有底物中第k种底物所占的比例。
2.2.5 肠道微生物群落演替的总动力学模型
基于以上速率方程,采用自下而上的耦合方法建立肠道微生物群落演替的总动力学模型,它由M+m个一阶非线性常微分方程构成的状态方程机理模型:
根据式(1)、(2)、(3)和(4),第i种微生物种群动态变化的一阶动力学方程表示为:
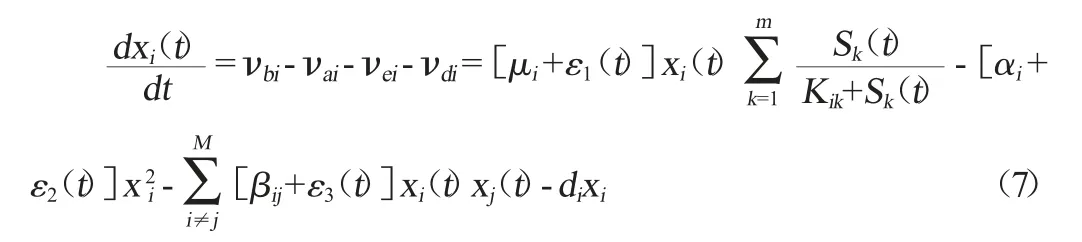
根据式(1)、(5)和(6),第k种底物浓度变化的一阶动力学方程表示为:
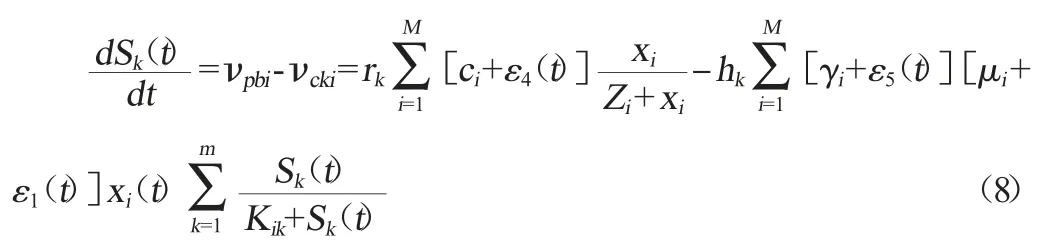
因为肠道微生物群落演替模型式(7)和式(8)中的各个参数都有明确的生物学意义,它们与微生物的基因型密切相关,因此第i种微生物可以表示为一个参数向量[μi,αi,βij,Kik,di,ci,γi,Zi],而第k种底物可以表示为[rk,hk],这两个参数反映了底物自身及其对微生物的特性。
在肠道微生物群落演替动力学模型的基础上,根据实验数据,通过非线性最小二乘法和遗传算法对模型中参数进行了有效地辨识,结果如表1所示。

表1 微生物群落演替动力学模型中的参数Table 1 Parameters in microbial community succession dynamics models

续表
2.3 肠道微生物群落演替过程的Monte Carlo实验
本研究通过数学模型,模拟产生了20~50种微生物,5~15种可直接利用的基质,在进行数值仿真之前,根据本研究问题的复杂性、求解精度和收敛速度等,对仿真的方法进行了优化设置,结果见表2。
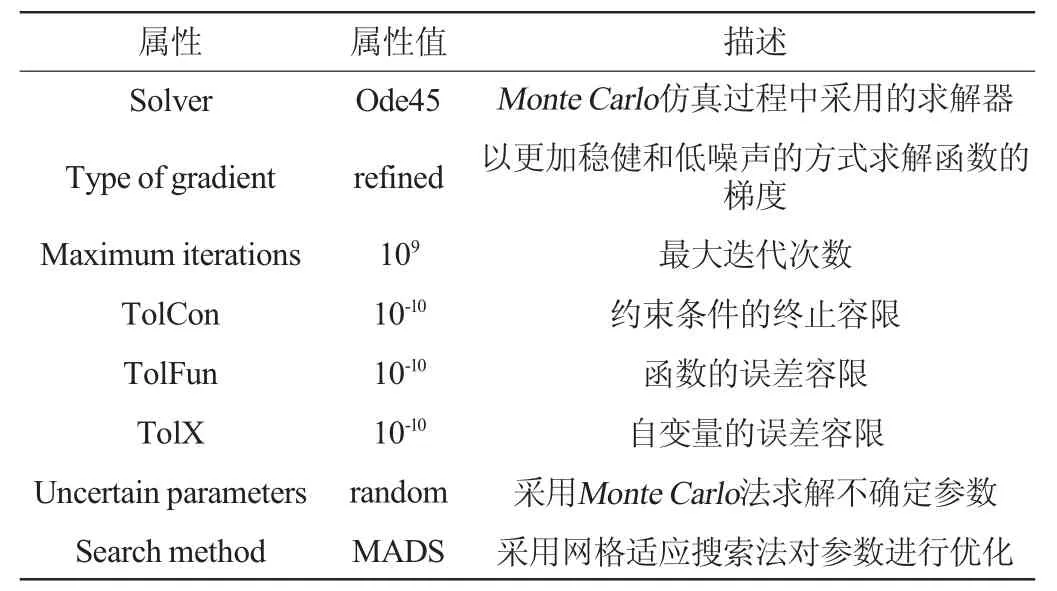
表2 Monte Carlo仿真实验的设置Table 2 Settings of Monte Carlo simulation experiment
由于微生物群落在人体肠道内的演替模型(式(7)、式(8))是非线性的,不存在解析解,因此本研究在Matlab/Simulink平台上建立它的计算机模型,通过Monte Carlo仿真获得模型的数值解,模拟肠道微生物群落的演替过程[17]。当模型(式(7)、式(8))中表示环境扰动的参数(ε1~ε5)的设置为表1中较大功率时,得到的结果见图4。

图4 长期饮酒组肠道微生物群落演替的基本模式Fig.4 Basic pattern of gut microbial community succession in long-term alcohol consumption group
由图4可知,在长期饮酒干预下,肠道中的微生物种群数量经过一段时间混沌状态的瞬态响应后,微生物群落达到低物种多样性的稳定状态,达到平台期的微生物种群呈现出稳定的弱相关波动,即波动的自相关性和互相关性均大大减小,这说明在瞬态响应过程中,大量的微生物被竞争排斥掉,只有少数微生物物种可以稳定共存[18]。
然而当模型(式(7)、式(8)中表示环境扰动的参数(ε1~ε5)的设置为表1中较小功率时,得到结果见图5。由图5可知,空白对照组肠道中的微生物种群数量一直在正常的环境背景扰动下,呈现出随机波动的特征,微生物群落达到高物种多样性的非稳定平衡状态--周期或非周期波动,达到平台期的微生物种群呈现出强相关的波动,即波动的自相关性和互相关性均大大增加,避免了强烈的竞争排斥,可以使大量微生物物种得以稳定共存。值得一提的是,通过计算机仿真充分证明了图4和图5呈现的微生物群落在人体肠道中的演替模式是相当稳健的,它对模型的初始条件和参数相当不敏感,演替模式具有良好的普适性[19]。
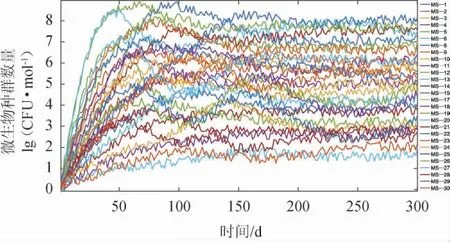
图5 空白对照组微生物群落演替的基本模式Fig.5 Basic pattern of gut microbial community succession in control group
由于对微生物来说长期饮酒是一种逆境因子[20],根据对生态系统扰动的频率、强度和严重程度,长期饮酒在肠道可以被视作一种对微生物群落的中度扰动[21-23]。生态学中的中度扰动假说结果表明,相比于对生态系统的低扰动或强扰动,中度扰动下可以有效地对生态系统的物种产生影响,既不灭绝也不零干扰[13]。从仿真结果中可以看到,微生物在演替开始时,多个种群几乎都呈指数增长,其增长速度决定于它们各自的最大瞬时增长率。然而随着肠道生态位真空逐渐被填满,各个种群实际增长率会越来越小,由于它们各自的增长率、竞争能力和环境容量不可能完全相同,因此必然会出现这样一个时刻,此时某些种群的实际增长率已下降为零,而另一些种群却在继续增长,成为优势种,这是竞争过程的转折点,对于优势种来说,它的继续增长不仅会强化对衰退种群的竞争排斥作用,而且会使衰退种群的实际增长率为负值。此时衰退种群已经开始下降,而增长种群仍继续扩大,这样下去,衰退种群就会走向灭绝,产生剧烈地竞争排斥现象,最后只有少数的优势种可以共存,大多数菌种都灭绝掉,无法形成与维持肠道微生物的多样性。
从统计学角度来看,肠道的微生物种群动态可以看作是一个随机过程。根据随机过程原理,如果对某一随机过程进行一次长时间的观测和记录,那么从这些时间序列的数据中,就可以得到该随机过程精确的统计特征,如过程的均值、方差、自相关和互相关函数,以及功率谱密度等。因此,观测和记录这5个菌属的相对丰度动态,4个月的监控是一个足够长的时间,在实验期间菌属的相对丰度动态可以遍历它所有可能的相对丰度值,所获得的时间序列可以精确地得到它们动态随机过程的统计特征[25]。
3 结论
本研究在微生物生态学理论、实验数据和观测现象的基础上,对长期饮酒者肠道微生物多样性形成和维持机制进行了研究。发现作为中度扰动因子,长期饮酒可以有效地加强微生物的随机生长响应,并伴随着强烈的微生物种内和种间竞争随机过程、基质的生产和消耗随机过程,它们有效地减弱了各个微生物种群波动的自相关性和互相关性,肠道菌群多样性降低且有紊乱的趋势。通过建立具有正态分布随机参数的系统动力学模型,在大量的Monte Carlo仿真实验基础上确证了研究结果。肠道内得到的微生物群落演替模式具有鲁棒性的,即它对模型的初值始和参数不敏感,所得到的微生物演替模式具有相当的普适性。本研究为理解饮酒对肠道微生物的生态学效应提供了理论基础,同时为酒精摄入性疾病防控建立科学依据。
猜你喜欢
中日友好医院学报(2022年4期)2022-10-15
数学物理学报(2022年4期)2022-08-22
数学物理学报(2021年4期)2021-08-30
数学物理学报(2021年4期)2021-08-30
北京航空航天大学学报(2021年7期)2021-08-13
教育周报·教育论坛(2020年3期)2020-10-21
看世界·学术下半月(2020年7期)2020-09-10
科学(2020年2期)2020-08-24
科技资讯(2018年16期)2018-10-26
广东农业科学(2017年10期)2018-01-25
