
Development of chloroplast marker for identif ication of Ulva species*
2023-01-04 03:03DahaiGAOQingchunZHANGZhongminSUN
Dahai GAO , Qingchun ZHANG , Zhongmin SUN
1 Key Laboratory of Exploration and Utilization of Aquatic Resources, Ministry of Education, Shanghai Ocean University,Shanghai 201306, China
2 Shanghai Engineering Research Center of Aquaculture, Shanghai Ocean University, Shanghai 201306, China
3 Department of Marine Organism Taxonomy and Phylogeny, Institute of Oceanology, Chinese Academy of Sciences (IOCAS),Qingdao 266071, China
4 Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences,Qingdao 266071, China
Abstract Large-scale Ulva-caused green tides has posed various environmental and ecological problems as well as economic consequences. It is important to identify them accurately and quickly. The current universal markers based on ITS, rbc L, and 5S spacer sequences were defective to distinct closely related species in the genus of Ulva. In this study, by investigating the intergenic regions of chloroplast (cp) genome,a novel marker of c15 (based on ycf 3- rps 7) were reliable for discrimination of Ulva species. Notably, the resolution of c15 was suitable for resolve the closely related species of U. linza and U. prolifera, which may provide tools for deciphering the blooming mechanisms of green tides.
Keyword: Ulva; linza-procera-prolifera (LPP) complex; molecular makers; ycf 3- rps 7 region
1 INTRODUCTION
As a type of harmful algal boom, green tide caused byUlvaspp. is a source of marine pollution (Blomster et al., 2002). Recently, large-scale green tides occur frequently in coastal areas worldwide (Smetacek and Zingone, 2013). In China,Ulvagreen tides occurred in the Bohai Sea, Yellow Sea, and South China Sea in last decade, which damaged local marine environments and ecological systems (Zhang et al., 2019; Xie et al.,2020b). Specif ically, the world’s largest green tide in the Yellow Sea have lasted for 15 consecutive years since 2007, whose causal species were identif ied asU.prolifera(Pang et al., 2010; Zhang et al., 2011,2018, 2019; Zhao et al., 2012; Qi et al., 2016).Based on laboratory culturing analysis, it showed that the multiple life cycle modes were exhibited forU.prolifera, involving sexual reproduction with bif lagellate gametes and quadrif lagellate meiospores,and asexual reproduction from bif lagellate spores,quadrif lagellate spores, and unfertilized gametes(Liu et al., 2015; Zhang et al., 2017). AlthoughU.proliferacan be recognized from its related taxa in adult stage, it is hard to morphologically discriminate each another in the young stage even for the experts.Phylogenetically,U.prolifera,U.procera, andU.linzacould not be distinct and formed a clade called linza-procera-prolifera (LPP) clade based on solo ITS orrbcL markers (Shimada et al., 2008, 2016). And the hybridization studies indicated thatU.proliferaandU.linzawas the closely relatedUlvaspecies with complex speciation history, as the marine strains ofU.proliferawere complete reproductive isolated withU.linzaexhibiting by gamete incompatibility(Hiraoka et al., 2011), whereas some brackish strains ofU.proliferashowed partial gamete compatibility withU.linza(Xie et al., 2020a).
To solve the relationships among closely related species inUlva, several molecular approaches were reported. Besides the combination of ITS orrbcL markers, Shimada et al. (2008) f irstly introduced the 5S rDNA spacer sequences to analyze the genetic diversity within LPP complex, and this marker was widely used for studying origin of f loatingU.proliferaunder intra-specif ic level (Zhao et al., 2019).However, the using of 5S rDNA marker has some def iciencies as the PCR products of 5S rDNA marker were not specif ic as several bands were observed during electrophoretic analysis (Liu et al., 2022). On the other hand, as 5S rDNA marker was from nuclear genome, it will confuse the analysis if specimens were heterozygous. In another study, a PCR-based marker of sequence characterized amplif ied region(SCAR) was designed for identifying theU.proliferapopulations (Zhao et al., 2015), but the application of SCAR marker is limited as only f loatingU.proliferacould be specif ically amplif ied. In addition, the ribosomal large subunit (LSU) and intergenic spacer(IGS) sequences inU.proliferawere investigated,but their ability to delimitate closely related species remains uncertain (Shen et al., 2019). Therefore,specif ic markers should be exploited to identify the closely related species in a stable manner.
In land plants, the molecular markers have developed from chloroplast genome, including genecoding genes ofrbcL andmatK, as well as intergenic regions oftrnH-psbA andtrnL-F, which were widely used for species delimitation and phylogenetic analysis (Mishra et al., 2016). However, in algae, few studies were involved in the application of molecular markers from chloroplast (cp) genome. In this study,by investigating the intergenic regions of cp genome,we tried to develop novel marker with purpose to discrimination ofUlvaspecies.
2 MATERIAL AND METHOD
2.1 Sampling and species identif ication
The specimens ofUlvaspp. used in this study were collected from the coast and off shore areas of China,which distributed in ecologically diverse locations(Fig.1). For morphologically identif ication, the healthy adult individuals were selected and washed several times with sterile seawater to remove surface attachments, and then herbarium comparation were carried out according to the taxonomic features ofUlvataxa. Voucher herbarium specimens were deposited in the Marine Biological Museum of the Chinese Academy of Sciences (MBMCAS) (Fig.2).For molecular identif ication, the genome DNA of each specimen were extracted and ITS barcoding were characterized via PCR amplif ication, sequencing, and Blast analysis.
2.2 DNA extraction, PCR amplif ication, and sequencing
Total genomic DNA was extracted from fresh specimens with a DNeasy Plant kit (Tiangen Biotech.Co. Ltd., Beijing, China) following manufacturer’s instructions. The ITS region was amplif ied by primers designed forUlvataxa by Hayden et al. (2003) (F:5ʹ-TCTTTGAAACCGTATCGTGA-3ʹ; R: 5ʹ-GCTTATTGATATGCTTAAGTTCAGCGGGT-3ʹ), and the c15 region of cp genomes were designed in this study (F: 5ʹ-CTGACATACCATCACGATAGT-3ʹ;R: 5ʹ-CGTATTATTTCACCAGACCCTT-3ʹ). The PCR reaction were carried out by TaKaRa Ex Taq enzyme in 25-L reaction column (TaKaRa, Shiga,Japan), with condition of an initial denaturation at 94 °C for 5 min, 36 cycles at 94 °C for 50 s, 54 °C for 50 s, and 72 °C for 1 min, and a f inal elongation step of 10 min at 72 °C. All PCR products were examined by electrophoresis on a 1% agarose gel and then sequenced using autosequencer (ABI,3730) according to manufacturer’s instructions (ABI,BigDye®Terminator v3.1 Cycle Sequencing Kit).
2.3 Data analysis
To develop high-resolution markers, the cp genomes ofU.prolifera(KX342867),U.linza(KX058323),andU.f lexuosa(KX579943) were recruited for comparative genomic analysis. The sequences alignment of cp genomes was analyzed and the conserved regions were identif ied by mVISTA online software (https://genome.lbl.gov/vista/mvista/submit.shtml). Thus, several less conserved regions were selected for primers design and evaluated via routine PCR and sequencing analysis. For phylogenetic analysis, the sequences were aligned using Clustal module in MEGA software v.11 (Tamura et al.,2021) and then manually adjusted. The phylogenetic trees based on ITS andycf3-rps7 sequences were constructed with Neighbor-joining (NJ), maximum likelihood (ML), and Bayesian inference (BI) methods,respectively. NJ analysis was carried out by MEGA software v.11 with 1 000 bootstrap replicates, whereas ML and BI analysis were carried out by PhyloSuite software (Zhang et al., 2020) following corresponding instructions. Trees were visualized using FigTree v.1.4.2 (http://tree.bio.ed.ac.uk/software/f igtree/).

Fig.1 Sampling of Ulva species
3 RESULT
3.1 Chloroplast genomes comparison and marker development

Fig.2 The specimens of representative species in the genus of Ulva
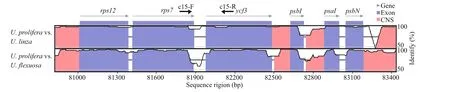
Fig.3 Comparative analysis of the chloroplast genome of three Ulva species
Based on the cp genomes alignment ofUlvaprolifera,U.linza, andU.f lexuosa, a high degree of conservation of synteny was exhibited, indicting the evolutionary relationships of these species was closely related (Fig.3). It is shown that most coding regions were highly conserved, ref lecting the evolutionary conservation of these genes to the function of chloroplast. In addition, there were conserved no-coding sequences (CNS) scattered in the alignment, suggesting these sequences might have conserved regulatory roles to the genes.Therefore, the less conserved sequences located in intergenic region could be theoretically used to develop molecular markers. Accordingly, to PCRamplify these intergenic regions, dozens of primers were designed based onU.proliferagenomic sequences. After evaluation by gel electrophoresis and sequencing, it is showed that majority of primer pairs were failed to obtain a specif ic and unique PCR product. Nevertheless, a specif ic PCR product corresponding to the intergenic region ofrps7 andycf3 genes could be stabilized amplif ied by one primer pair labelled c15, exhibiting a potential role as novel molecular marker.
3.2 Sequence analysis of ycf 3- rps 7 region in Ulva taxa
To evaluate the usability ofrps7-ycf3 region, the sequence divergence between species was analyzed.The corresponding amplif ication regions of primer pair c15, which include intergenic sequences ofrps7-ycf3, were retrieved from 9 publishedUlvacp genomes. According to the sequence alignment results, the coding region were highly conserved(Fig.4). Specif ically, there were two highly variable regions (HVR1 and HVR2) located in the intergenic regions, suggesting these sequences divergence could be used for markers to distinguish species.

Fig.4 Sequence alignment of rps 7- ycf 3 regions of representative Ulva cp genomes
3.3 Discrimination of U. linza and U. prolifera through phylogenetic analysis
To validate the resolution of the c15 marker, the specimens collected from the coastal area of China were used for phylogenetic analysis (Table 1).These specimens could be identif ied as eightUlvaspecies based on their distinguished morphological characteristics (Fig.2).U.linzaandU.proliferacould not separate as monophyly in the ITS tree,which clustered into LPP complex as previous studies(Fig.5a). However, in the c15 tree,U.linzaandU.proliferasplit into two separated clades clearly,suggesting the high resolution of this marker (Fig.5b).Furthermore, the monophyly of other sixUlvaspecies were unaff ected, which was consistent with ITS orrbcL markers.
4 DISCUSSION
The key to control theUlvagreen tides is to identify the causal species accurately, which is important to prevent the occurrence of bloom. Several studies have demonstrated that theU.proliferaon bamboos,nets and ropes of aquaculture infrastructure were the seed source to green tides in the Yellow Sea (Liu et al., 2013, Zhang et al., 2017). As there were four major species ofU.linza,U.compressa,U.f lexuosaandU.proliferabeen identif ied inNeopyropiacultivation area (Zhang et al., 2011), it is diffi cult to morphologically discriminate each another in the young germlings. Thus, the early interference by recognizing theU.proliferaspecif ically will increase the effi ciency of controlling green tides blooms.
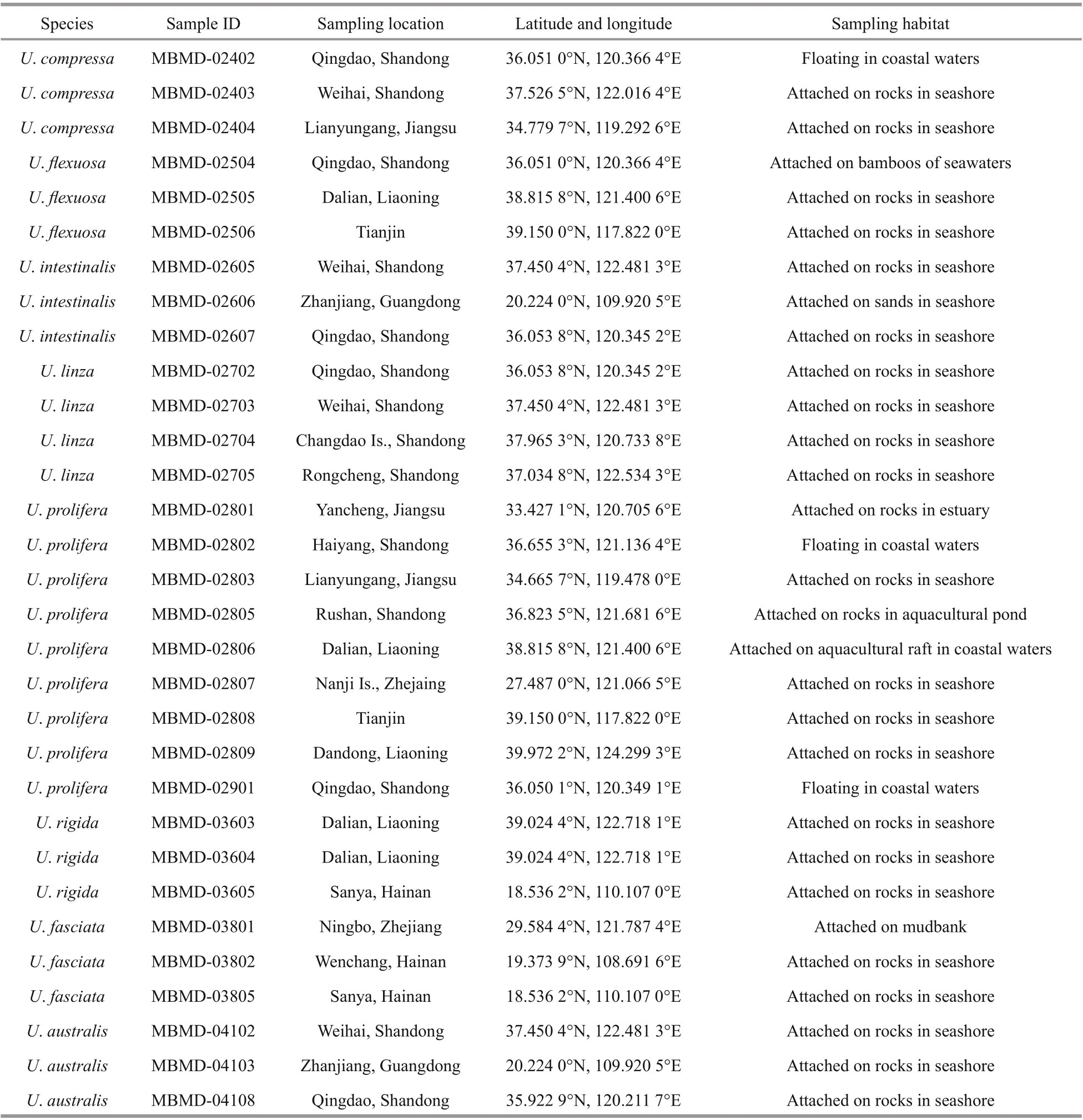
Table 1 Samples information used in the study
In this study, a novel marker was developed from cp genome for phylogenetically discriminating ofUlvaspecies in a high-resolution manner.Specif ically, comparing with tradition markers, c15 could precisely resolve the species relationships ofU.linzaandU.proliferain LPP complex (Fig.5b).Although the nuclear marker 5S rDNA were applied to distinct the species in LPP complex (Shimada et al., 2008, 2016), the specif icity of this marker was doubted as incomplete concerted evolution (Liu et al., 2020). Moreover, the new c15 marker could be used for other frequently distributedUlvaspecies,indicating its potential to work as universal barcode to investigate the cryptic biodiversity of this taxa.
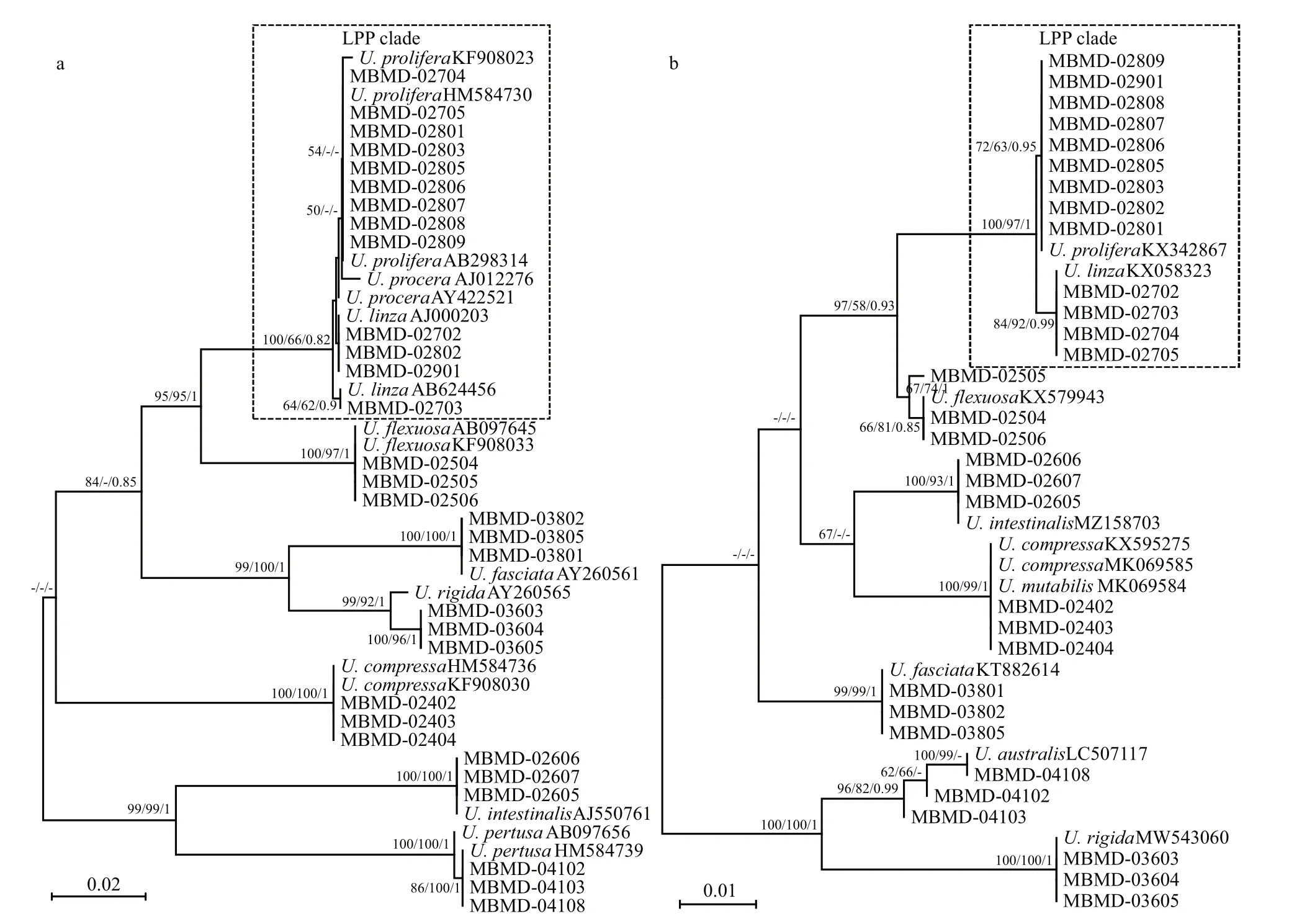
Fig.5 Phylogenetic trees based on ITS (a) and rps 7- ycf 3 (b) sequences
The identif ication of closely related species by molecular method was challengeable as the events of hybridization and gene f low between species occur frequently. As a result, diff erent strategies are selected to solve problematic genera when resolution is desired.In most cases, the molecular markers developed from organelle genomes are uniparental inherited and single copy, providing resolution up to inter or intraspecies levels (Lahaye et al., 2008; Matiz-Ceron et al., 2022). For example, the markers ofatpF-atpH andtrnH-psbA can simultaneously discriminate all the 46 representative medicinal plant species of 28 families (Thakur et al., 2019). However, given the diff erent evolutionary pattern and reproductive mode between land plant andUlvaspecies, whether the cp genome ofUlvawas inherited uniparentally need to be investigated.
In our analysis, the interspecif ic relationships were not completely identical between ITS and c15 phylogenies. The reasons for this inconsistent could be the diff erent evolutionary rate and patterns of distinct markers (Wang et al., 2017). As both ITS and c15 are from no-coding intergenic region, it is suitable to identify taxa but not analyze phylogenetic relationships. Therefore, emerging phylogenomic analysis combining protein-coding markers could expected to better understanding the phylogenetic relationships forUlvaspecies. Besides, the intraspecif ic diversif ication inUlvaspecies were not well-solved, which need novel markers been developed.
5 CONCLUSION
In this study, by investigating the intergenic regions of chloroplast (cp) genome, a novel marker of c15 (based onycf3-rps7region) were reliable for discrimination ofUlvaspecies. Notably, the resolution of c15 was suitable for resolve the closely related species ofU.linzaandU.prolifera, which may provide tools for deciphering the blooming mechanisms of green tides.
6 DATA AVAILABILITY STATEMENT
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
 Journal of Oceanology and Limnology2022年6期
Journal of Oceanology and Limnology2022年6期
- Journal of Oceanology and Limnology的其它文章
- Overview of harmful algal blooms (red tides) in Hong Kong during 1975–2021
- Information standardization for typical toxic and harmful algae in China’s coastal waters—a case study of Karenia mikimotoi*
- Biochemical composition of the brown tide causative species Aureococcus anophageff erens cultivated in diff erent nitrogen sources*
- Identif ication of paralytic shellf ish toxin-producing microalgae using machine learning and deep learning methods*
- Screening for lipophilic marine toxins and their potential producers in coastal waters of Weihai in autumn, 2020*
- First observation of domoic acid and its isomers in shellf ish samples from Shandong Province, China*