
Diversity and community structure of eukaryotic microalgae in surface sediments in the central Bohai Sea, China, based on a metabarcoding approach*
2023-01-04 03:03ZhaohuiWANGChaofanWANGMaotingWANGWeiguoLIWencongZHONGLeiLIUTaoJIANG
Zhaohui WANG , Chaofan WANG , Maoting WANG , Weiguo LI , Wencong ZHONG ,Lei LIU , Tao JIANG
1 College of Life Science and Technology, Jinan University, Guangzhou 510632, China
2 Engineering Research Center of Tropical and Subtropical Aquatic Ecological Engineering, Ministry of Education, Guangzhou 510632, China
3 School of Ocean, Yantai University, Yantai 264005, China
Abstract Sediment samples were collected at 17 stations in the central Bohai Sea, China, and the diversity and community structure of eukaryotic microalgae were assessed by metabarcoding the V4 region of 18S rDNA. A total of 930 operational taxonomic units (OTUs) were detected for microeukaryotes, including 98 algal OTUs. The algal communities comprised 42 genera belonging to 19 classes of six phyla, and they were dominated by chrysophytes and dinof lagellates. Dinof lagellates were also the most diverse microalgal group.The nano-sized dinof lagellates Biecheleria halophila and Azadinium trinitatum occurred abundantly in the study area; however, they have not been reported previously, as they may be overlooked or misidentif ied in light microscopy. Many pico-sized chlorophytes were detected in the sediment samples. Sixteen of the detected OTUs were assigned to potentially harmful and/or bloom-forming microalgae, suggesting some potential risks of harmful algal blooms in the central Bohai Sea. The capacity of metabarcoding to detect morphologically cryptic and small species makes this method a suffi ciently sensitive means of detection for assessing eukaryotic microalgae in sediments.
Keyword: microalgae; dinof lagellate cysts; high throughput sequencing; marine sediment; the Bohai Sea;resting stages
1 INTRODUCTION
Eukaryotic microalgae are important primary producers of marine ecosystems and regarded as the basis of food web structures (Worden et al., 2015).They have diverse nutritional types, including phototrophic, heterotrophic, mixtrophic, and parasitic(reviewed by Brodie et al., 2017). The diversity and community structure of marine microalgae play an important role in global biogeochemical cycles, the stability of marine ecosystems, and the mitigation of global climate change (Martinez et al., 2009; Vogt,2015). However, some microalgae produce toxins and/or form harmful algal blooms (HABs), which could have negative eff ects, causing severe economic losses to f isheries, aquaculture, and tourism activities,and exerting major environmental and human health eff ects (Anderson et al., 2002). Of all phytoplankton,approximately 150 species are categorized as harmful microalgae in the online version of the IOC Taxonomic Reference List of Toxic Plankton Algae (Lundholm et al., 2009 onwards). Those algal species, especially toxin-producing algae, could harm other marine organisms and human health through the food chain,such as f ish and shellf ish.
Many planktonic microalgal species have been reported to produce dormant stages as a part of their life cycles (Head, 1996; Von Dassow and Montresor,2011) in the forms of resting spores and resting cysts(hereafter referred to as resting stages). The formation of resting stages is prompted by adverse environmental conditions, nutrient limitation (Kremp et al., 2009), or pressure from grazing (Anderson and Rengefors,2006). Once the resting stages arrived, they sink to the sea f loor and become a part of the benthic assemblages (Head, 1996). Resting stages have resistant cell wall, and they could survive in bottom sediments for long periods (decades to 100 years;reviewed by Ellegaard and Ribeiro, 2018).Germination of resting stages occurs if they are resuspended in the water column and exposed to appropriate light intensity, temperature, nutrients, and most importantly, oxygen availability (McQuoid,2002). Resting stages are regarded as the “seed bank”of algal blooms, and they play an important role in initiating HABs (Smayda, 2002; Genovesi et al.,2009).
The identif ication and quantif ication of the resting stages of eukaryotic microalgae have traditionally been based on microscopic observations and cell counting after separating cells from sediments. These methodologies for separation and observation have been developed over decades; however, some limitations and uncertainties exist, including losses during separation and concentration, biases in the recovered groups, and diffi culties associated with morphological identif ication (Piredda et al., 2017).Microscopic observations require professional taxonomic expertise for distinguishing the tiny morphological diff erences among species (Shang et al., 2019). Furthermore, only a small proportion of phytoplankton resting stages is known for their corresponding vegetative cells and vice versa.Misidentif ication is thus common and almost unavoidable due to these diffi culties (Gómez et al.,2017; Shang et al., 2019).
Advances in the sequencing of DNA extracted from natural water and sediment samples off er huge opportunities for biodiversity monitoring and assessment, particularly where the collection or identif ication of whole organisms is impractical(reviewed by Lear et al., 2018). DNA sequence analysis using metabarcoding has been widely applied in characterizing the biodiversity and community structure of microeukaryotes in marine sediments(reviewed by Lear et al., 2018; Gran-Stadniczeñko et al., 2019). This method provides information on many organisms affi liated with several trophic levels in a biological system (Lanzén et al., 2016), some of which are diffi cult to identify with classic methods,hard to culture, fragile, and rarely analyzed (Egge et al., 2015). Metabarcoding off ers the prospect for determining their distribution in the world’s oceans and enables a more complete assessment of anthropogenic eff ects on ecosystems (Lekang et al.,2020), particularly for unicellular microalgae that often lack distinctive morphological characteristics(Vaulot et al., 1989) and have complex life cycles(Von Dassow and Montresor, 2011; Decelle et al.,2012).
In the past decade, numerous studies have analyzed benthic microeukaryotic community structure and its response to environmental changes. Metabarcoding has been successfully applied to track temporal changes in eukaryotic communities in coastal sediments (Salonen et al., 2019), assess the distribution and diversity of marine eukaryotes (Fonseca et al.,2014; Wu and Huang, 2019), monitor the eff ects of f ish farming and human activities on benthic communities (Pochon et al., 2015; Stoeck et al.,2018), reveal modern and past eukaryotic communities(Garcés-Pastor et al., 2019), and predict the ecological conditions of estuaries and coastal sea areas (Chariton et al., 2015). In addition, DNA metabarcoding has a great potential for the assessment of phytoplankton resting stages in environmental sediments(Dzhembekova et al., 2018; Liu et al., 2020).
The Bohai Sea, China’s only inland sea, is a semiclosed, shallow marginal sea in Northeastern China.With the increase in industrialization and urbanization in the Bohai Sea region in recent decades, large quantities of industrial and domestic wastewater have been discharged into the sea and pose a serious environmental risk to the ecosystem (Zhu et al.,2020). As the Bohai Sea exchanges only limited quantities of water with the open sea, it has become one of the most polluted sea areas in China (Duan and Li, 2017). Furthermore, algal blooms, especially those of HABs, have recently become more frequent in the Bohai Sea (Xu et al., 2017). Over the last two decades, a number of studies analyzing environmental conditions and the pollution status in the Bohai Sea have been published (Wei et al., 2019; Yang et al.,2019; Zhu et al., 2020). However, the resting stages of eukaryotic communities in sediments from the Bohai Sea have been rarely revealed. Only a few studies described the distribution of benthic diatoms (Wang et al., 2020) and cysts of toxic dinof lagellates (Dai et al., 2020) in the Bohai Bay. In addition, most of these studies have been based on microscopic observations,which may have not allowed species identif ication for many resting stages.
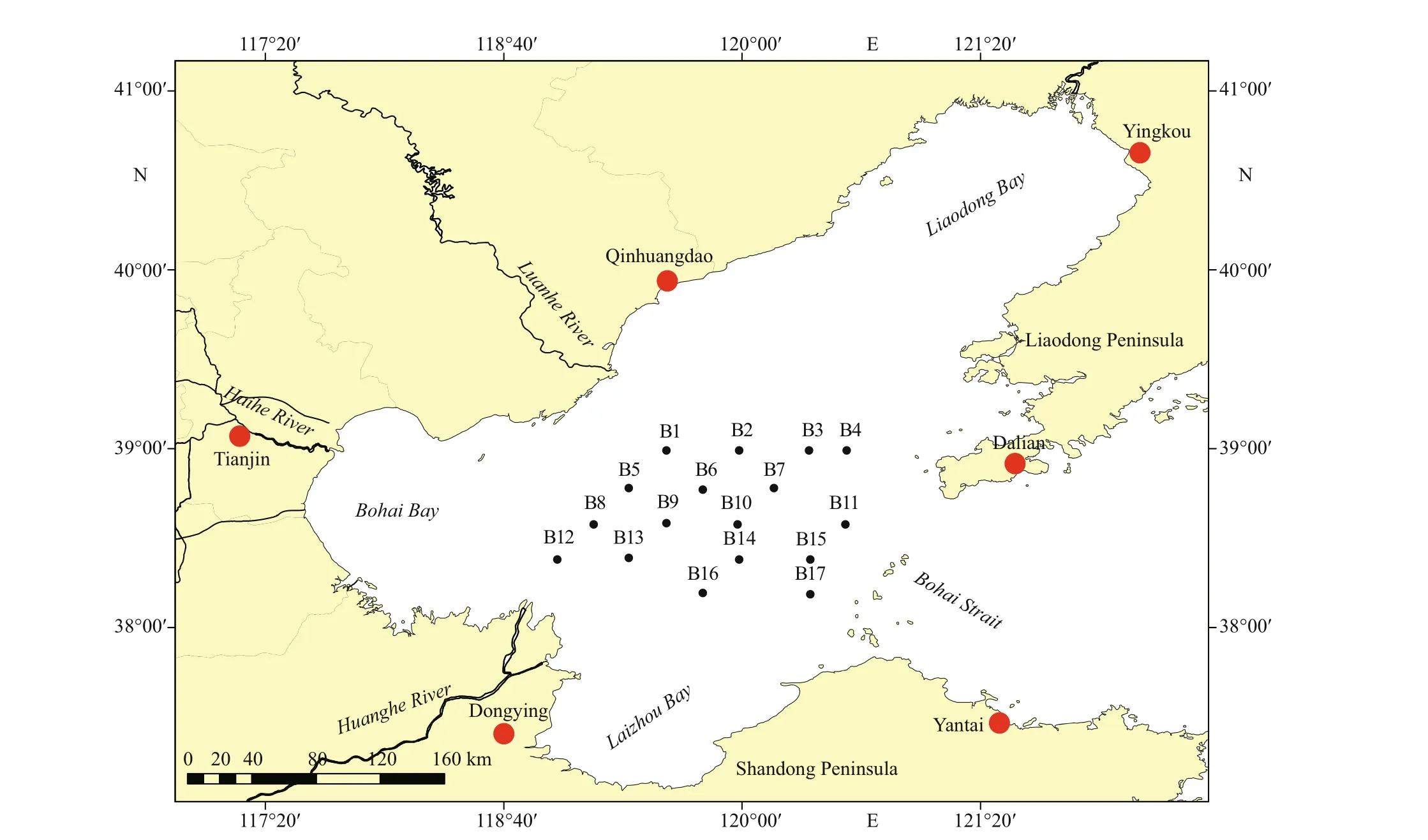
Fig.1 Map of the central Bohai Sea, showing major cities (red dots), rivers, and sampling stations (black dots) in this study
In this study, high-throughput sequencing (HTS)was used to analyze the biodiversity and distribution of eukaryotic microalgae in surface sediments from the central Bohai Sea. This study aimed to assess the diversity of benthic microalgae by metabarcoding,with a focus on the phytoplankton resting stages; to understand the distribution of HAB species; and to discuss the potential for HABs in the central Bohai Sea.
2 MATERIAL AND METHOD
2.1 Study area and sediment collection
The Bohai Sea is a shallow semi-enclosed epicontinental sea. It includes four parts, i.e., Bohai Bay, Laizhou Bay, Liaodong Bay, and central Bohai Basin (Fig.1). The area of the Bohai Sea is 77 000 km2,and the average water depth is 18 m. Many rivers,including the Huanghe (Yellow) River, Haihe River,Luanhe River, and Liaohe River, f low into the Bohai Sea (Fig.1). The study area is located in the central Bohai Sea.
Surface sediment samples were collected by a Van-Veen Grab with a 10-cm×10-cm frame at 17 stations in the central Bohai Sea in May 2015 (Fig.1). Samples of the upper surface sediments (ca. 2 cm in depth)were collected in triplicate at each station with a polyethylene scraper. Before DNA extraction, the sediment samples were stored under dark conditions at low temperatures (4–8 °C) for 6 months to inactivate most of the microalgal vegetative cells and to ensure that the algal DNA extracted from the sediment samples mostly belonged to the resting stages (Piredda et al., 2017).
2.2 DNA extraction, PCR amplif ication, and sequencing
Environmental DNA was extracted and purif ied from 0.5-g sediment samples by using the DNeasy PowerSoil Kit (Qiagen GmbH, Germany) following the manufacturer’s protocols (Liu et al., 2020). PCR amplif ication of the V4 region of 18S rDNA was performed using universal eukaryotic primers 3NDf 5ʹ-GGCAAGTCTGGTGCCAG-3ʹ (Cavalier-Smith et al., 2009) and V4_euk_R2 5ʹ-ACGGTATCTATCTCTTCG-3ʹ (Bråte et al., 2010)with lengths of ca. 450 bp. PCR products were sequenced using the Illumina MiSeq PE300 (San Diego, CA, USA) platform by Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China.
2.3 Operational taxonomic unit (OTU) analysis and taxonomic assignment
Raw sequence reads were denoised, trimmed, and f iltered using the UPARSE (v8.0.1623) algorithm of USEARCH (Edgar, 2013) and the Ribosomal Database Project (RDP, version 2.2, http://sourceforge.net/projects/rdp-classifer, Caporaso et al., 2010).OTUs were generated based on 97% similarity after removal of singletons and doubletons. Identif ication of each OTU was inferred using the RDP classif ier against the SILVA 18S rRNA database (release138,www.arb-silva.de). The eukaryotic algal OTUs were conf irmed based on taxonomic information with AlgaeBase, a global algal database (Guiry and Guiry,2020, https://www.algaebase.org/). The raw data were deposited into the National Center for Biotechnology Information Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra) with the accession number PRJNA722689.
2.4 Statistical analysis
MAFFT version 7.475 (Katoh and Standley, 2013)was used to align the eukaryotic algal DNA sequences in this study. A maximum-likelihood tree was constructed with FastTree version 2.1.11 software(Price et al., 2009) with default parameters by using the GTR + CAT substitution model for 1 000 bootstrap replicates. The phylogenetic tree was annotated using the packages ggtree (Yu et al., 2017) and Adobe Illustrator 2020. The spatial distribution of eukaryotic algal reads was illustrated by inverse distance weighting (IDW) interpolation (weighting power of 2.0), and a sampling map and IDW interpolation map were produced by ArcMap 10.2.
3 RESULT
3.1 Sequencing statistics and diversity estimates
A total of 813 046 reads of eukaryotic rDNA were generated from 17 surface sediment samples from the central Bohai Sea after removal of sequence errors and chimeras. The number of eukaryotic sequences per sample ranged from 30 821 reads to 72 429 reads,with an average of 48 809 reads (Fig.2a). The dominant length ranged from 441 bp to 462 bp, with an average of 446 bp. At 97% sequence identity, 930 OTUs were detected, and 144–418 OTUs were recorded per sample, with an average of 302 OTUs(Fig.2b). Among all eukaryotic sequences, 126 679 reads (15.58%) and 98 OTUs (10.54%) were assigned to the eukaryotic microalgae. The numbers and relative abundances of algal reads ranged between 383 and 33 174 reads and between 0.89% and 81.49%(Fig.2a), with averages of 7 452 reads and 17.01%,respectively. The algal DNA reads were numerous at stations B4 and B12, exceeding 30 000 reads and contributing >70% of the total reads. On the contrary,the algal OTU reads were low at stations B9, B13,B16, and B17, at which fewer than 1 000 algal reads were recorded. Algal OTU richness was detected in 17–58 OTUs (Fig.2b), and it was high at stations B2,B3, B5, and B11 (>50 OTUs). The proportions of algal OTU richness to eukaryotic richness were comparable among samples (10.4%–16.0%).
3.2 Community structure of eukaryotic microalgae
The algal communities were composed of 42 genera belonging to 19 classes of six phyla (i.e.,Chlorodendrophyceae, Chloropicophycea,Mamiellophyceae, Prasinophyceae,Pyramimonadophyceae, and Trebouxiophyceae in Chlorophyta; Bacillariophyceae, Coscinodiscophyceae,Mediophyceae, Chrysophyceae, Dictyochophyceae,Pelagophyceae, and Raphidophyceae in Ochrophyta;Dinophyceae, Syndiniophyceae, and Noctilucophyceae in Dinophyta; Katablepharidophyceae in Cryptophyta;Kinetoplastea in Euglenozoa; and Pavlovophyceae in Haptophyta). Sixty algal OTUs (61.22%) were identif ied at the genus level, and only 42 OTUs (42.86%)were fully identif ied at the species level. Altogether, 39 species were detected in this study (Supplementary Table S1). Some algal OTUs were rare taxa, with 29 OTUs represented by fewer than 20 reads and 23 OTUs represented by fewer than 10 reads. Among these 29 OTUs (<20 reads), 12 OTUs were identif ied at the species level (Supplementary Table S1).
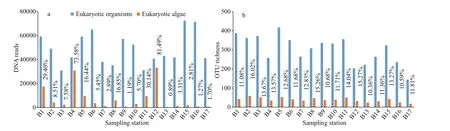
Fig.2 DNA reads (a) and OTU richness (b) of eukaryotic organisms and eukaryotic algae in surface sediments from the central Bohai Sea
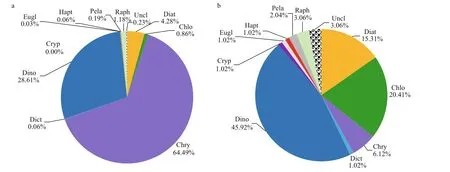
Fig.3 Percentage composition of DNA reads (a) and OTU richness (b) of microalgae at the phylum or class level
Figure 3 shows the percentage composition of DNA reads and OTU richness of microalgae at the phylum or class level. Chrysophyceae dominated the eukaryotic algal communities, contributing 64.49% of the algal sequences (Fig.3a). However, only six chrysophyte OTUs were detected in this study, four of which were only identif ied at the class level. Two chrysophyte OTUs (OTU757 and OTU10) contributed 52.64% and 11.63% to the algal sequences,respectively. Dinophyta (dinof lagellates, including the three classes in Dinophyta) was the second most abundant algal group (28.61%). The common dominant dinof lagellates includedBiecheleriahalophile(Biecheler) Moestrup, Lindberg &Daugbjerg (OTU511),AzadiniumtrinitatumTillmann& Nézan (OTU624),Scrippsiellaacuminata(Ehrenberg) Kretschmann, Elbrächter, Zinssmeister,Soehner, Kirsch, Kusber & Gottschling (OTU354),andPolykrikoshartmanniiZimmermann (OTU62),which contributed 28.58%, 23.66%, 3.91%, and 1.76% of the dinof lagellate sequences, respectively. In addition, one Syndiniales OTU (OTU234) and two unclassif ied dinof lagellate OTUs (OTU366 and OTU498) occurred abundantly, contributing 12.05%,12.24%, and 9.51% to dinof lagellate sequences,respectively. Diatoms (Bacillariophyceae,Coscinodiscophyceae, and Mediophyceae) contributed to 4.28% of the total algal sequences.ThalassiosiraCleve (OTU927) predominated in diatoms,contributing 86.56% to diatom reads. The sequences of raphidophytes contributed 1.18%, and the others contributed less than 1%. Dinof lagellates were the most diverse microalgal group (Fig.4b), and they represented 45.92% of OTU richness (45 OTUs),followed by chlorophytes (20 OTUs, 20.41%), diatoms(15 OTUs, 15.31%), and chrysophytes (6 OTUs,6.12%). The other groups included only 1–3 OTUs.
Dinof lagellates dominated at 12 stations (Fig.4a),with sample relative abundance ranging between 1.96% and 98.13% and averaging 61.60%.Chrysophytes predominated in f ive samples, with percentages of 0.09%–97.88% per sample and 23.50% on average. The relative abundances of diatoms ranged between 0.03% and 38.87%, with an average of 10.16%. Raphidophytes occurred abundantly at station B11 (13.66%), and they were less abundant in other samples. Dinof lagellates were the most diverse group in almost all samples (Fig.4b),with 7–28 OTUs (27.45%–53.19%) per sample. OTU richness was high for chlorophytes and diatoms, with 2–18 (8.70%–35.29%) and 2–11 (3.70%–23.53%)OTUs in each sample, respectively.
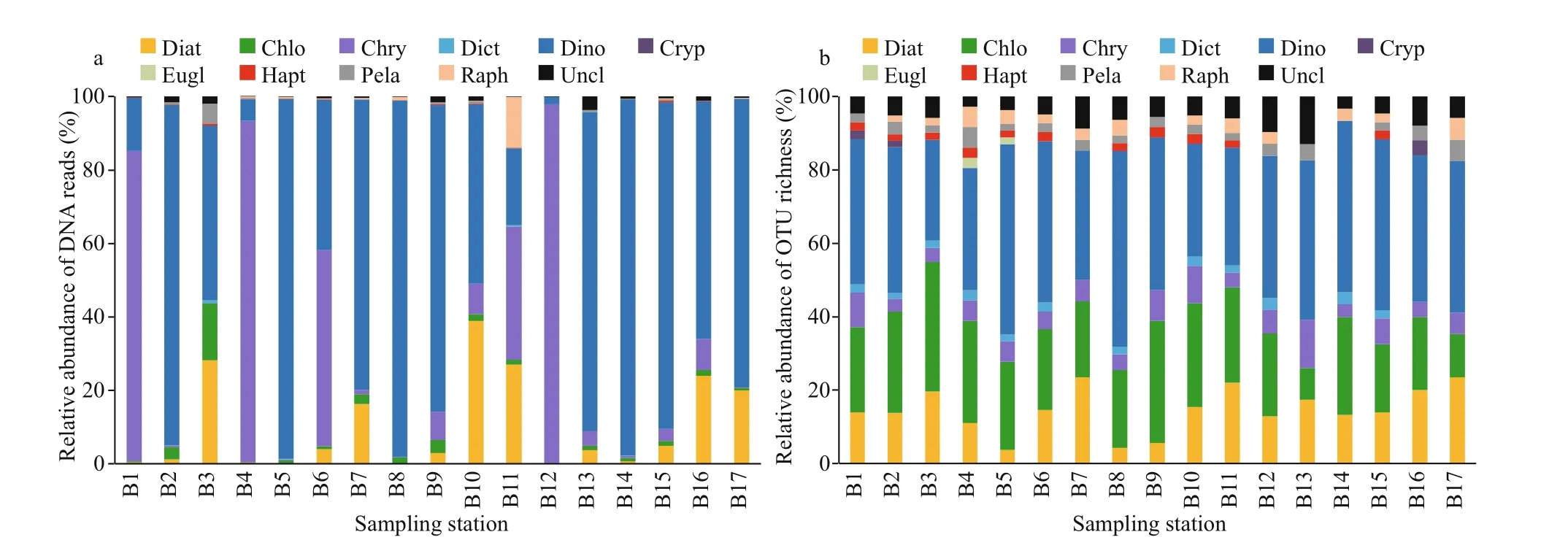
Fig.4 Relative abundance of DNA reads (a) and OTU richness (b) of eukaryotic algae at the phylum or class level in 17 stations
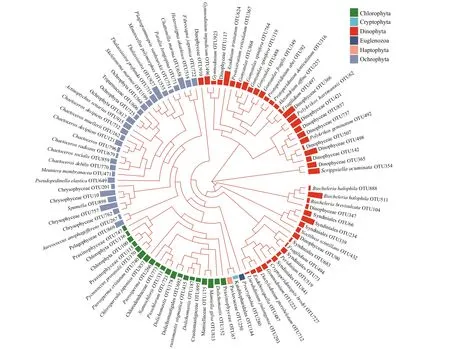
Fig.5 Maximum-likelihood phylogenetic tree (FastTree) of the algal OTUs from this study belonging to the six phyla of eukaryotic algae
A maximum-likelihood tree was constructed to compare the relationships among the 98 algal OTUs(Fig.5). The OTUs in Haptophyta, Cryptophyta,Chlorophyta, and Ochrophyta were clustered into a large clade. The OTUs in Ochrophyta and Chlorophyta were separately clustered together into a large subclade. One OTU in Cryptophyta (OTU144) and one OTU in Haptophyta (OTU250) were grouped together with low bootstrap support. One OTU in Euglenozoa was ungrouped. The phylogenetic relationships among OTUs in Dinophyta were more diverse, and they included f ive subclades and one ungrouped OTU. In addition, not all OTUs in the same genus were clustered together, and some OTUs in the same genus or order were distributed in diff erent subclades. For example, two OTUs ofFragilidiumBalech ex Loeblich III were distinct from each other,three OTUs ofGymnodiniumStein were separately grouped into two subclades, and the OTUs in Syndiniales were grouped into two close subclades.Non-monophyly of genera may be caused by misidentif ication due to incorrect sequence names deposited in the SILVA database. They could also suggest the high genetic diversity and polymorphism in Dinophyta and ref lect the hypervariability of the V4 region of 18S rDNA.
3.3 Distribution of dominant and harmful algal bloom species
Sixteen of the detected OTUs were assigned to harmful and/or bloom-forming microalgae, including eight taxa in Dinophyceae (Alexandriumaffi ne(Inoue& Fukuyo) Balech,Azadiniumtrinitatum,Gonyaulaxspinifera(Claparède & Lachmann) Diesing,Noctilucascintillans(Macartney) Kofoid & Swezy,Polykrikoshartmannii,Polykrikosgeminatus(Schütt) Qiu &Lin,Protoceratiumreticulatum(Claparède &Lachmann) Bütschli, andScrippsiellaacuminate),four diatom taxa (ChaetocerosdebilisCleve,ChaetocerosdecipiensCleve,ChaetocerossocialisLauder, andSkeletonemamarinoiSarno & Zingone),one taxon in Pelagophyceae (Aureococcusanophageff erensHargraves & Sieburth), and three in Raphidophyceae (Chattonellamarina(Subrahmanyan) Hara & Chihara,FibrocapsajaponicaToriumi & Takano, andHeterosigmaakashiwo(Hada) Hada ex Hara & Chihara). Some of these harmful species belonged to rare taxa, whose sequences were less than 20 reads, includingAlexandriumaffi ne,Noctilucascintillans, andAureococcusanophageff erens(Supplementary Table S1). However, some occurred widely and abundantly,such asAzadiniumtrinitatum,Scrippsiellaacuminata,Chattonellamarina, andPolykrikoshartmannii.
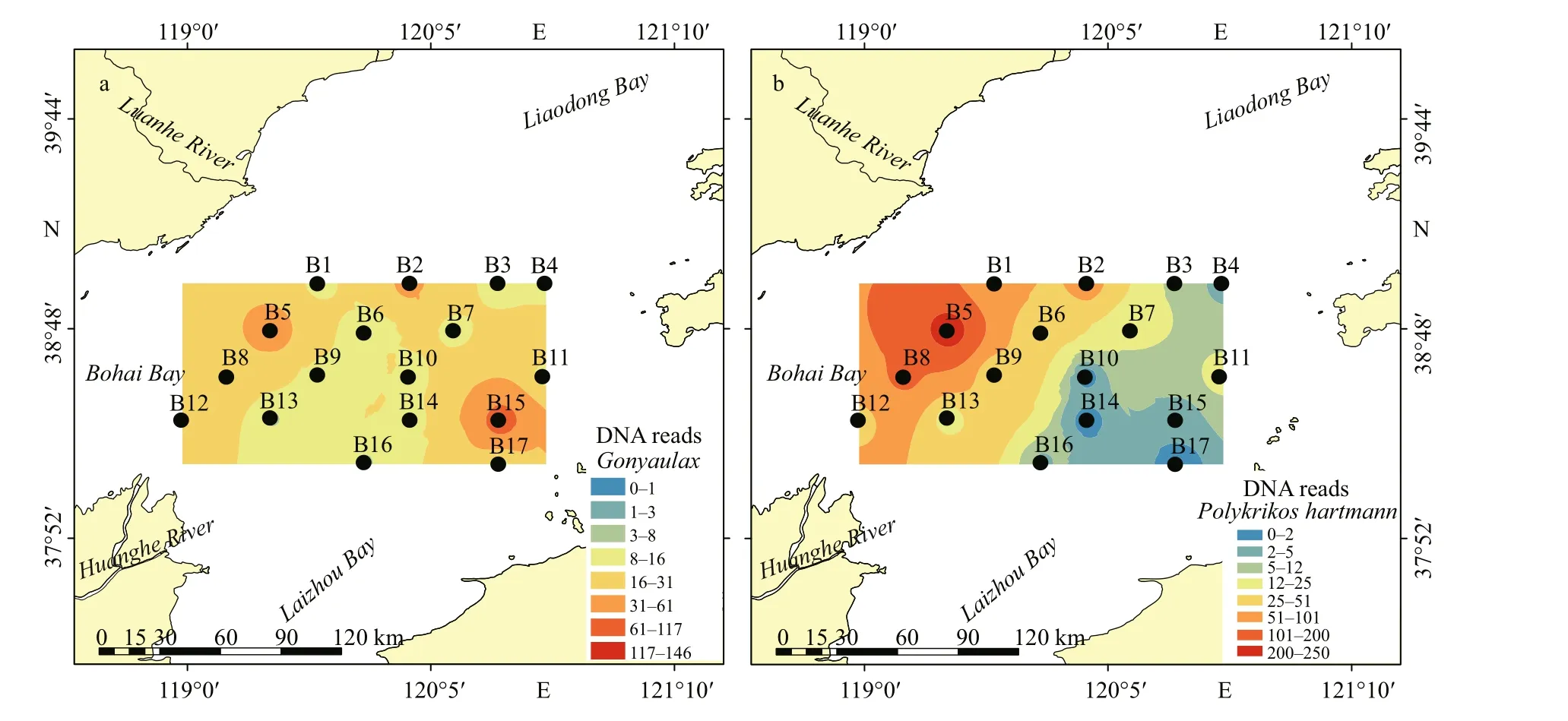
Fig.6 Distribution of DNA reads of dominant and potentially harmful/toxic microalgae in surface sediments from the central Bohai Sea

Fig.6 Continued
Many species inGonyaulaxDiesing could produce yessotoxin (YTX), includingGonyaulaxspinifera(Howard et al., 2009) andGonyaulaxtayloriiCarbonell-Moore (Álvarez et al., 2016). Five OTUs ofGonyaulax(OTU119, OTU488, OTU349, OTU764,and OTU868) were obtained (Supplementary Table S1), two of which were assigned toGonyaulaxspinifera, and one of which was identif ied asGonyaulaxfragilis(Schütt) Kofoid.Gonyaulaxsequences ranged from 0 to 146 reads, with the highest reads at station B15 (Fig.6a). Another YTX producer,Protoceratiumreticulatum(Howard et al., 2009), only occurred at station B5 (Supplementary Table S1).Polykrikoshartmanniiis an ichthyotoxin producer(Tang et al., 2013) that was detected at 15 stations and prevalent at station B5 and the surrounding stations(Fig.6b).AlexandriumHalim is an important source of paralytic shellf ish poisoning (PSP, Hallegraeff , 2003).Only one OTU ofAlexandriumwas detected in this study (Alexandriumaffi ne, OTU 257), and it occurred at three stations (B4, B14, and B15) with low sequence numbers (Supplementary Table S1).
Some species of the planktonic dinof lagellate genusAzadiniumElbrächter & Tillmann, such asAzadiniumspinosumElbrächter & Tillmann andAzadiniumpoporumElbrächter & Tillmann, produce azaspiracids (AZAs, Tillmann et al., 2014). OneAzadiniumOTU (OTU624), which was the nontoxicAzadiniumtrinitatum, was detected in this study(Supplementary Table S1). The abundance ofAzadiniumtrinitatumwas higher in the northwestern area, particularly at stations B1 and B2 (Fig.6c).Scrippsiellaacuminata,Polykrikosgeminatus, andNoctilucascintillansare common HAB species in coastal waters (Hallegraeff , 2003).Scrippsiellaacuminatawas found at almost all stations(Supplementary Table S1). Sequences ofScrippsiellaacuminatapeaked at station B5 in the southwestern corner, with high levels in western waters (Fig.6d).Polykrikosgeminatuswas found at only three stations(Supplementary Table S1).Noctilucascintillansappeared at only a few stations, with very low numbers of reads (Supplementary Table S1).BiecheleriaMoestrup, Lindberg & Daugbjerg (mostlyBiecheleriahalophile), a thin-walled nanosized dinof lagellate, was widely and abundantly distributed(Supplementary Table S1). High reads ofBiecheleriaoccurred in the northwestern area, with the highest at station B5 (Fig.6e).
Toxic and harmful algal species other than dinof lagellates mostly occurred in the east, with the highest reads at station B11 (Fig.6f–h). Many species in Raphidophyceae could produce ichthyotoxins(Basti et al., 2016). Three OTUs of raphidophytes were detected in the present study (Supplementary Table S1), and they were identif ied asChattonellamarina(OTU658),Fibrocapsajaponica(OTU722),andHeterosigmaakashiwo(OTU518).Chattonellamarinawas distributed at eight stations, with higher reads at station B11 (Fig.6f).FibrocapsajaponicaandHeterosigmaakashiwooccurred in low sequence numbers (Supplementary Table S1).
Thalassiosirawas the most abundant diatom genus in this study, and two of its OTUs were detected(Supplementary Table S1).ChaetocerosEhrenberg is a common genus of planktonic diatoms that form resting spores, and seven OTUs ofChaetoceroswere identif ied in this study; however, all OTUs had low sequence numbers (Supplementary Table S1). Higher sequence numbers ofThalassiosiraandChaetocerosoccurred in the western sea area, with the highest at station B11 (Fig.6g–h). Another algal bloom species,Skeletonemamarinoi, occurred at eight stations but in very low sequence numbers (Supplementary Table S1).
Two Pelagophyceae OTUs were detected in this study, including the brown tide speciesAureococcusanophageff erens, which only occurred at two stations(B2 and B4) with very low sequence numbers(Supplementary Table S1).
4 DISCUSSION
4.1 Community structure of eukaryotic algae
A total of 98 algal OTUs from 17 surface sediment samples were detected from the central Bohai Sea in this study. Xu et al. (2017) derived 409 OTUs of the eukaryotic phytoplankton community from samples of the coastal waters of the Bohai Sea, within which 75 OTUs were identif ied at the species level. The OTU richness of phytoplankton was far higher than that of eukaryotic algae in the sediments. Most phytoplankton vegetative cells are generally thought to not be able to survive in sediments for a long time,and only resting stages of phytoplankton are well preserved in sediments after long-term cold-dark storage (reviewed by Ellegaard and Ribeiro, 2018). In the present study, the sediment samples were stored in a dark place at 8 °C for 6 months before DNA extraction to ensure that the algal DNA extracted from sediments was mostly from resting stages(Piredda et al., 2017). Therefore, the algal OTU richness in sediments is generally lower than that obtained from the water column, because only a small component of phytoplankton could form resting stages (Head, 1996; McQuoid and Hobson, 1996).
Most taxa identif ied at the species level in this study have been reported to form resting stages(Supplementary Table S1). However, whether the other taxa could form cysts was not known. For example,Azadiniumtrinitatumoccurred abundantly in sediments from the central Bohai Sea(Supplementary Table S1) and the southern Chinese coasts (Liu et al., 2020), suggesting that it is a cystforming species. Indeed, cyst formation is an important characteristic in genusAzadinium(Tillmann et al., 2016). The wide and abundant occurrences of some parasitic taxa, such as the parasitic Syndiniales(OTU234), were probably due to the presence of their hosts. In addition, the dark cold treatment before DNA extraction could not exclude the fragmental DNA released from the dead cells. These DNA fragments should generally be represented as rare OTUs according to Shang et al. (2019). The rare OTUs with 1–2 reads (singletons and doubletons)were removed in the present study. Therefore, the algal sequences we detected in this study mostly existed as resting stages.
Dinof lagellate cysts constitute an important component of sedimentary assemblages of microalgal resting stages (Ellegaard and Ribeiro, 2018). In addition, high numbers of DNA copies (up to tens or hundreds of thousands of copies per cell) have been found in dinof lagellates (Lin, 2011) and thus may be over-represented in sequencing (Liu et al., 2017;Chen et al., 2021). Dinof lagellates have been also found to make substantial contributions to DNA reads and OTU richness in the phytoplankton communities in the Bohai Sea (Xu et al., 2017), though diatoms have constituted the predominant phytoplankton group represented in microscopic observations (Guo et al., 2014; Wang et al., 2019). The DNA reads and OTU richness of diatoms only ranked the third among the eukaryotic algae in the present study. Resting spores have been reported only in some centric diatoms, and they are seldom reported in pennate species (McQuoid and Hobson, 1996). Diatom OTUs obtained from sediment metabarcoding are mostly a few resting stages forming species, such as the centric diatomsChaetoceros,SkeletonemaGreville, andThalassiosira(Montresor et al., 2013; Piredda et al.,2017; Dzhembekova et al., 2018). Therefore, the diversity and abundance of diatoms are usually underestimated in sediment metabarcoding studies.Quantif ication discrepancies of taxa are a common problem of the current approaches used for metabarcoding, and some experts suggested that presence/absence should be used in metabarcoding instead of relative abundance data (Buchner et al.,2019; Bailet et al., 2020).
The high abundance of chrysophyte sequences was accounted for by the predominance of some particular OTUs at some stations, including OTU757 at stations B4 and B12 and OTU10 at station B1. The results agreed with the f indings of previous studies on eukaryotic algae and germinated phytoplankton in sediments from the South China Sea, where chrysophytes occurred predominantly at some particular stations (Liang, 2018; Liu et al., 2020).Cyst formation is a characteristic feature of chrysophytes, and several chrysophyte isolates were observed to form cysts under laboratory conditions(Findenig et al., 2010). Although the dominant chrysophyte OTUs could not be identif ied at the species level in the present study, the wide and abundant occurrence of these sequences suggested their ubiquitous distribution and cyst-formation characteristics.
Diverse chlorophytes (green algae) were notably detected in this study, some of which occurred widely and abundantly (Fig.5 and Supplementary Table S1),includingMamiellagilva(Parke & Rayns) Moestrup(Mamiellophyceae) andPterospermacristatumSchiller (Pyramimonadophyceae). Xu et al. (2017)found a wide diversity of chlorophytes, including f ive species in Mamiellophyceae, two in Nephroselmidophyceae, and two in Pycnococcaceae,in the phytoplankton community in the coastal waters of the Bohai Sea based on 18S rDNA sequencing.MamiellagilvaandPterospermacristatumwere regarded as previously unreported species in the Bohai Sea. Tragin and Vaulot (2018) analyzed the Ocean Sampling Day metabarcoding dataset, which provides sequences from the V4 region of 18S rDNA for 157 samples collected at 143 coastal stations, and they found the chlorophytes to be ubiquitous and dominant in coastal waters, especially those in Mamiellophyceae. In the present study,Mamiellophyceae was also the most diverse and abundant chlorophyte class. Most chlorophyte taxa detected in this study were pico-sized and diffi cult to sample and distinguish in traditional phytoplankton surveys. In addition, green algae are ubiquitous in nature, and they are likely to be introduced during DNA extraction and sequencing. However, the dominant green algae in this study were all marine species, and they occurred widely in the sediments.Few reports of resting stages in the prasinophyte green algaePyramimonasgelidicolaMcFadden,Moestrup & Wetherbee (Van den Hoff et al., 1989)andMantoniellaDesikachary (Ellegaard et al., 2016)could be found. The wide and abundant distribution of these marine green algae in benthic (present study)and planktonic communities (Xu et al., 2017) suggests their perennial occurrence in the Bohai Sea.
4.2 Occurrence of small-sized species and HAB species
The small thin-walled dinof lagellateBiecheleriahalophilawas the most dominant dinof lagellate OTU in this study, and it was present at all stations(Supplementary Table S1).Biecheleriais a group of nanosized phytoplankton taxa, most of which could form resting cysts (Takahashi et al., 2014). Species ofBiecheleriahave rarely been reported in routine phytoplankton surveys due to their small sizes and thin walls (Luo et al., 2013; Takahashi et al., 2014).Based on electron microscopy and HTS techniques,diverseBiecheleriaspecies have been reported(Takahashi et al., 2014). The cysts and motile cells ofBiecheleriaare widely distributed in coastal sea areas worldwide (Takahashi et al., 2014; Dzhembekova et al., 2018; Salonen et al., 2019). Luo et al. (2013)reported the f irst occurrence ofBiecheleriaspecies(Biecheleriacincta(Siano, Montresor & Zingone)Siano) in China after sediment incubation.Biecheleriacinctahas also been found to occur abundantly and widely in sediments from the southern Chinese coasts(Liu et al., 2020). However,Biecheleriahalophilahas not yet been reported in the Chinese sea areas in either planktonic or benthic communities. The wide distribution of this species suggests that it could be a common dinof lagellate in the Bohai Sea; however, it may be ignored during routine phytoplankton investigations.
Another nanosized dinof lagellate,Azadiniumtrinitatum, occurred at all stations in high abundance(Supplementary Table S1).Azadiniumtrinitatumis a newAzadiniumspecies recently described in the North Atlantic by Tillmann et al. (2014). Many species ofAzadiniumare responsible for AZA shellf ish poisoning (AZP); however, no known AZAs were found inAzadiniumtrinitatum(Tillmann et al.,2014). ThoughAzadiniumpoporum, which causes AZP, has been previously reported to be widely distributed along the Chinese coasts (Gu et al., 2013;Liu et al., 2020), the present study was the f irst to reportAzadiniumtrinitatumin the Chinese sea areas.The cells ofAzadiniumare generally small (ca. 15 μm in length and 10 μm in width) and rather inconspicuous under light microscopy. Determination of the tiny diff erences in morphological characteristics requires electron microscopy or tedious high-resolution light microscopy (Tillmann et al., 2014). Identif ication ofAzadiniumfrom f ixed f ield samples is thus problematic and further complicated by other species with similar sizes and shapes (Tillmann et al., 2014). The wide distribution ofAzadiniumalong the Chinese coasts(this study, Gu et al., 2013; Luo et al., 2013; Liu et al.,2020) and worldwide in sea areas (Akselman and Negri, 2012; Tillmann et al., 2014) indicates a global distribution of this genus (Tillmann et al., 2014).
Among all microalgal taxa, 16 potentially algal bloom-forming species were detected in this study(e.g., PSP-causingAlexandriumaffi ne; YTX producersGonyaulaxspiniferaandProtoceratiumreticulatum; ichthyotoxicPolykrikoshartmannii,Chattonellamarina,Fibrocapsajaponica, andHeterosigmaakashiwo; and bloom-causing species,such asNoctilucascintillans,Polykrikosgeminatus,Scrippsiellaacuminate,Chaetocerosspp.,Skeletonemamarinoi, and the brown tide speciesAureococcusanophageff erens). These taxa may produce toxins harmful to humans or form blooms associated with mortality of f ish or birds in marine coastal waters (Grattan et al., 2016). Some of these harmful algal taxa and other OTUs were detected in very few sequences (<20 reads), includingAlexandriumaffi ne,Noctilucascintillans, andAureococcusanophageff erens(Supplementary Table S1). These taxa were detected in very small amounts in this study, suggesting that they rarely occur in nature or were the outcome of biases in the analysis(Chariton et al., 2015; Gran-Stadniczeñko et al., 2019;Bailet et al., 2020).
HABs have frequently occurred in the coastal sea areas of the Bohai Sea starting from the late 1990s(Song et al., 2016).Aureococcusanophageff erens,PhaeocystisglobosaScherff el,Heterosigmaakashiwo,Kareniamikimotoi(Miyake & Kominami ex Oda) Hansen & Moestrup,Margalef idiniumpolykrikoides(Margalef) Gómez, Richlen &Anderson,Noctilucascintillans, andSkeletonemacostatum(Greville) Cleve have been the most frequently occurring bloom species (Song et al.,2016). Brown tides caused byAureococcusanophageff erenshave recurred frequently in the Northwestern Bohai Sea since 2009 (Huang et al.,2020). Most of these blooms occurred in coastal sea areas of the Bohai Bay due to the luxuriant nutrients discharged from the terrestrial sources (Song et al.,2016). However, resting stages formed after the nearshore blooms may be carried by water masses and settled into the deeper bottom sediments of the central Bohai Sea. These resting stages may germinate into vegetative cells and release them to the upper water column when the environmental conditions are suitable, which has the potential to form algal blooms in nutrient-rich waters. Therefore, the diverse HAB species recorded in this study and the high DNA reads of some particular species, such asAzadiniumtrinitatum,Scrippsiellaacuminata,Chattonellamarina, andPolykrikoshartmannii, suggests the potential risk of HABs in the central Bohai Sea.
5 CONCLUSION
The obtained environmental metabarcoding data improved the understanding of the composition of the microalgal community in surface sediments from the central Bohai Sea. Eukaryotic algae were dominated by chrysophytes and dinof lagellates. Dinof lagellates were the most diverse microalgal group. The identif ication of 16 potentially toxic and/or algal bloom species, some of which were found at all stations, highlighted the ecological signif icance of the benthic resting stages for HAB events. Some small taxa, such asBiecheleriahalophilaandAzadiniumtrinitatum, occurred widely and abundantly in sediments, indicating that they are common dominant species in phytoplankton communities; however, they may have been ignored in previous surveys relying on microscopic observations. The capacity of metabarcoding to detect morphologically cryptic species and better detect rare taxa makes this method suffi ciently sensitive to assess the composition of benthic microalgae in sediments.
6 DATA AVAILABILITY STATEMENT
The data that support the f indings of this study are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA)(http://www.ncbi.nlm.nih.gov/Traces/sra) with the accession number PRJNA722689.
 Journal of Oceanology and Limnology2022年6期
Journal of Oceanology and Limnology2022年6期
- Journal of Oceanology and Limnology的其它文章
- Overview of harmful algal blooms (red tides) in Hong Kong during 1975–2021
- Information standardization for typical toxic and harmful algae in China’s coastal waters—a case study of Karenia mikimotoi*
- Biochemical composition of the brown tide causative species Aureococcus anophageff erens cultivated in diff erent nitrogen sources*
- Identif ication of paralytic shellf ish toxin-producing microalgae using machine learning and deep learning methods*
- Screening for lipophilic marine toxins and their potential producers in coastal waters of Weihai in autumn, 2020*
- First observation of domoic acid and its isomers in shellf ish samples from Shandong Province, China*