
米曲霉RIB40高效同源重组和尿苷/尿嘧啶营养缺陷型菌株的构建
2023-01-03 07:13高育青张豪杰张丹凤潘裕添
食品工业科技 2023年1期
高育青,张豪杰,张丹凤,潘裕添,张 峰,
(1.闽南师范大学福建省菌类活性物质工程技术研究中心,福建漳州 363000;2.闽南师范大学生物科学与技术学院,福建漳州 363000)
米曲霉(Aspergillus oryzae)是公认的食品安全菌株,主要用于酱油和酒类的酿制,以及发酵产生蛋白酶、淀粉酶、脂肪酶和果胶酶等[1-4]。米曲霉3.042和米曲霉RIB40分别是在我国和日本广泛应用于食品工业的两种米曲霉菌种。它们的基因组长度都约为38 Mb,两者的大部分功能基因都比较保守,但在菌株生长、耐盐、环境抗性和次生代谢产物相关的基因方面具有一些不同点[5-6]。研究表明,米曲霉3.042菌株含有更多与细胞生长和环境抗性相关的基因而具有更强的生长活力。米曲霉RIB40菌株因含有较多的Na+、K+、甘油代谢相关基因,而更能耐受盐胁迫,使其在食品工业应用中更有价值[7]。此外,3.042菌株和RIB40菌株在醇、酸和氨基酸形成相关的酶的基因上也有差异,这些差异造成两者在生产酱油时产生不同的风味[8]。例如,侯丽华等[9]利用低盐固态工艺研究发现米曲霉RIB40菌株在原料溶出、蛋白质的分解以及生产还原糖方面的能力较强。王莹[10]在对两株菌株的发酵性能的比较中发现米曲霉RIB40发酵的酱油偏醇香,而米曲霉3.042偏酯香。本研究以米曲霉RIB40为出发菌株,构建遗传转化体系,能为我国食品工业提供更为多样的菌株选择。
借助遗传转化手段能在较短时间内实现基因的遗传改造,但首先需要建立高效的遗传转化体系。目前虽已有多种方法用于建立米曲霉的遗传转化体系,但是转化效率并不十分理想。Sun等[11]利用农杆菌法以pyrG基因作为筛选标记构建了尿苷/尿嘧啶营养缺陷型的米曲霉3.042遗传转化体系,但是只获得了0.1%的转化率。近年,毕付提等[12]同样利用农杆菌法以pyrG基因作为筛选标记,通过优化条件仅将米曲霉3.042的转化效率提高到了8.33%。刘雪[13]利用PEG-CaCl2介导的基因重组技术以米曲霉3.951为出发菌株构建的米曲霉pyrG单缺失菌株,结果发现其同源重组效率低于5%[14]。由于目前多数的遗传转化体系都是基于外源基因在出发菌株体内发生的基因重组,因此建立高效的遗传转化体系必须使出发菌株具有发生高效的基因重组的潜力。在构建的尿苷/尿嘧啶营养缺陷型菌株基础上,本研究进一步敲除了米曲霉中阻碍基因重组的Aoku70基因,能大大提高基因的重组效率。
本研究通过同源重组技术和5-氟乳清酸(5-Fluoroorotic Acid,5-FOA)的筛选作用对米曲霉RIB40的AopyrG和Aoku70基因进行双敲除,旨在建立具有高重组效率的遗传转化体系,为今后米曲霉RIB40的遗传改造提供了良好的出发菌株。
1 材料与方法
1.1 材料与仪器
米曲霉RIB40(Aspergillus oryzaeRIB40) 由华南理工大学潘力教授赠送;酵母提取物、蛋白胨英国OXOID公司;琼脂粉 BBI生命科学有限公司;吐温20、二甲基亚砜 西陇科学股份有限公司;Driselase、山梨醇、十二烷基硫酸钠 北京兰博利德生物技术有限公司;聚乙二醇4000 国药集团化学试剂有限公司;Tris 广州赛国生物科技有限公司;Lysing Enzymes和β-glucoronidase 美国Sigma公司;PDA培养基 美国BD公司;TransTaq® DNA Polymerase High Fidelity(HiFi) TransGen Biotech公司;2×Taq PCR Master Mix 北京金百特生物技术有限公司;琼脂糖凝胶回收试剂盒 上海惠凌生物技术有限公司;5-FOA 上海生物工程有限公司;所用YGT、复苏培养基及孢子洗脱液、DNA基因组小量抽提裂解液、原生质体裂解液、STC溶液和PEG Buffer的具体配方请参见文献[15]。
GF88DX全自动高压灭菌锅 厦门致微仪器有限公司;T30三槽基因扩增仪 杭州朗基科学仪器有限公司;Elix Essential5反渗透纯水系统 Merck Millipore;Synergy超纯水系统 Millipore;LAS600凝胶成像分析系统 美国GE公司;Nanodrop2000超微量分光光度计 Thermo Scientific;TGL-20M台式高速冷冻离心机 湘仪公司;Leica ICC50 E生物显微镜和LeicaSP8激光扫描共聚焦显微镜 徕卡公司;ZQLY-180F单目双层全温摇床 上海知楚仪器有限公司。
1.2 实验方法
1.2.1 米曲霉基因组DNA的提取 将菌丝或孢子液接种到YGT液体培养基中,于30 ℃培养2 d后,室温12000 r/min离心2 min收集菌丝。向菌丝中加入500 μL DNA基因组小量抽提裂解液,并于65 ℃金属浴裂解菌丝30 min。向裂解物中加入200 μL 3 mol/L乙酸钾,混匀后13000 r/min离心5 min。将上清液转移至另一个EP管中并加入等体积的异丙醇,混匀后12000 r/min离心10 min,收集沉淀物。向沉淀物中加入700 μL 75%的乙醇溶液洗涤,13000 r/min离心5 min后弃上清,于50 ℃烘箱中烘干沉淀,最后加30 μL无菌水溶解DNA。
1.2.2 基因片段的扩增 首先按照TransTaq® DNA Polymerase High Fidelity试剂盒的说明书对目的基因的上下游同源臂和标记基因进行PCR扩增,反应总体积为100 μL。对PCR产物用0.8%琼脂糖凝胶电泳并用琼脂糖凝胶回收试剂盒进行回收,用超微量分光光度计测定DNA浓度。接着将上下游同源臂和标记基因DNA片段等摩尔比混合,使用TransTaq®DNA Polymerase High Fidelity试剂盒进行重叠延伸PCR反应(Overlap Extension PCR,OEPCR)。OE-PCR反应体系按照TransTaq®DNA Polymerase High Fidelity试剂盒说明书进行,OE-PCR反应程序参照文献[16]。对OE-PCR产物用0.8%琼脂糖凝胶进行电泳并用琼脂糖凝胶回收试剂盒进行回收,用超微量分光光度计测定DNA浓度。
1.2.3 米曲霉原生质体的制备 将孢子液接种至100 mL YGT液体培养基中,30 ℃ 180 r/min培养16 h。用纱布收集菌丝,并用无菌水洗涤菌丝,再用滤纸吸干水分。向菌丝中加入5 mL原生质体裂解液,在30 ℃条件下裂解6 h。裂解后的混合物用擦镜纸过滤,并用预冷的1.2 mol/L山梨醇冲洗。将滤液于4 ℃冷冻离心机中4500 r/min离心15 min后,收集沉淀物。向沉淀物中加入300 μL预冷的1.2 mol/L STC溶液,再加入7%的二甲基亚砜重悬沉淀,分装后于-80 ℃保存。
1.2.4 米曲霉的转化及PCR验证 将琼脂糖凝胶回收的5 μg OE-PCR产物与预冷的200 μL STC溶液在无菌的EP管中混匀,再加入200 μL预冷的PEG Buffer混匀,最后加入100 μL原生质体。将混合物混匀后冰水浴30 min,然后再补加1 mL PEG Buffer,混匀后再冰水浴10 min。最后将上述混合液与顶层复苏培养基混匀后倒入已凝固的底层复苏培养基上,30 ℃培养至长出菌落。挑取转化子接种到YGT液体培养基中,培养2 d后,按照1.2.1中的方法提取DNA。转化子DNA的PCR验证条件按照2×Taq PCR Master Mix说明书进行。取5 μL PCR产物用1%琼脂糖凝胶进行电泳检测。
1.2.5AopyrG基因的敲除 从NCBI数据库中检索到米曲霉的乳清酸核苷-5’-磷酸脱羧酶基因(AopyrG)的序列,并设计AopyrG上下游引物(AopyrG-L1和AopyrG-L2,AopyrG-R1和AopyrGR2,表1)。按照1.2.2中的方法,以AopyrG上下游引物分别扩增AopyrG的上下游同源臂(AopyrGL、AopyrGR),然后以AopyrG-P1和AopyrG-P2引物(表1)通过OE-PCR将上下游同源臂融合成一个片段(AopyrGLR)。按照1.2.4中的方法,将AopyrGLR片段转化米曲霉RIB40的原生质体。在含有5-FOA培养基上挑取生长出来的菌株进行培养并进行PCR验证。
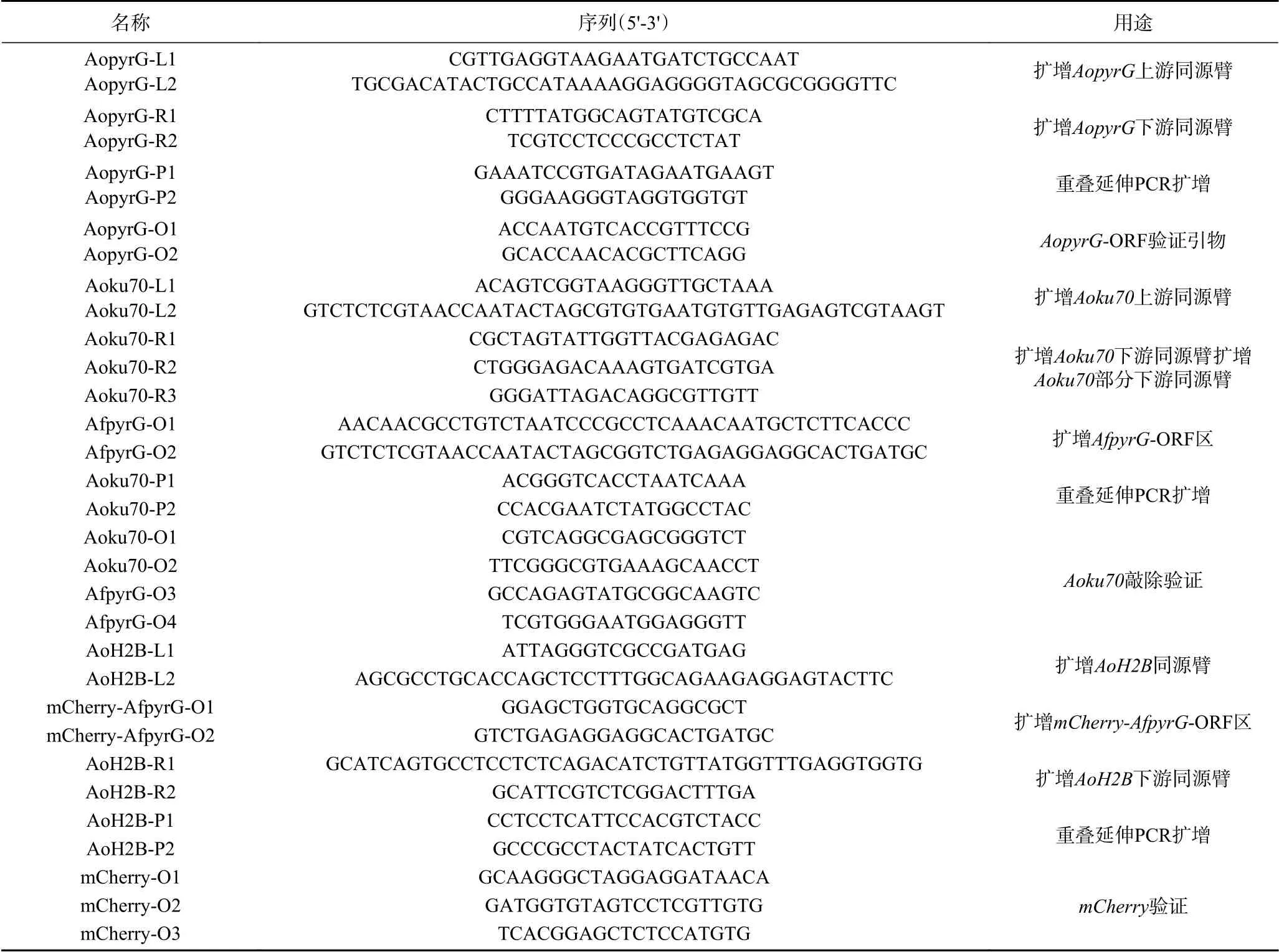
表 1 PCR扩增中所用到的引物Table 1 Primers used in PCR amplification
1.2.6Aoku70基因的敲除 从NCBI数据库中检索到米曲霉的Aoku70序列,并设计引物。按照1.2.2中的方法,以引物Aoku70-L1和Aoku70-L2,Aoku70-R1和Aoku70-R3,Aoku70-R1和Aoku70-R2(表1)分别扩增Aoku70上游(Aoku70L)、部分下游(Aoku 70r)和下游同源臂(Aoku70R),以引物AfpyrGO1和AfpyrG-O2(表1)扩增AfpyrG基因。先通过OE-PCR得到Aoku70r-AfpyrG片段,然后再通过OE-PCR得到Aoku70L-Aoku70r-AfpyrG-Aoku70R片段(LrGR)。按照1.2.4中的方法,将LrGR片段转化ΔAopyrG原生质体,挑取在不含有尿苷/尿嘧啶的培养基上生长出来的菌株进行培养和验证。将验证成功的菌株的孢子涂布到含有5-FOA的培养基上生长,挑取长出的菌株进行培养和PCR验证。
1.2.7AoH2B-mCherry核定位荧光菌株的构建从NCBI数据库中检索到米曲霉组蛋白AoH2B的基因序列,并设计引物。按照1.2.2中的方法,以引物AoH2B-L1和AoH2B-L2,AoH2B-R1和AoH2B-R2(表1)分别扩增AoH2B-ORF区及其上游同源臂(AoH2BL)、下游同源臂(AoH2BR),然后以引物mCherry-AfpyrG-O1和mCherry-AfpyrG-O2(表1)扩增mCherry-AfpyrG片段,最后以AoH2B-P1和AoH2B-P2引物(表1)通过OE-PCR将同源臂和mCherry-AfpyrG片段融合成一个片段(AoH2BmCherry-AfpyrG-AoH2BR)。按照1.2.4中的方法,将AoH2B-mCherry-AfpyrG-AoH2BR融合片段转化ΔAoku70ΔAopyrG双缺失菌株的原生质体,在复苏培养基上挑取生长出来的菌株进行培养及PCR验证。
1.2.8mCherry荧光的检测 将10 μL含有mCherry基因的米曲霉孢子液接种于YGT液体培养基中,在30 ℃摇床中培养。培养12 h后,12000 r/min 离心5 min收集萌发的孢子。将孢子分散铺于载玻片上,用10 μL无菌水冲洗孢子,并用擦镜纸小心地吸走水分,盖上盖玻片,在激光共聚焦显微镜下用激发波长587 nm,发射波长610 nm激发并观察红色荧光的位置。
1.2.9 米曲霉菌株生长状况的分析 将1 μL米曲霉孢子液接种于PDA培养基上,30 ℃培养6 d后用孢子洗脱液洗脱并收集孢子。用血球计数板在显微镜下进行孢子计数,然后用孢子洗脱液稀释孢子液至相同浓度。接种1 μL孢子液至PDA平板中央,30 ℃培养并每天测量菌落直径。
1.3 数据处理
菌落直径实验做3个平行实验,每个平行实验重复3次,以平均值和标准差表示数据结果。运用Excel和SPSS Statistics 23.0软件进行统计分析,采用独立样本T检验进行差异性比较,在95%显著性水平下分析差异的显著性,在Excel中进行柱状图分析。
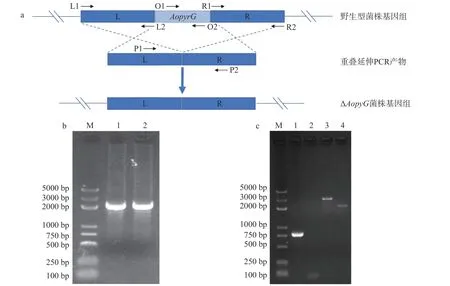
图 1 AopyrG基因敲除的验证Fig.1 Verification of the AopyrG gene knockout
2 结果与分析
2.1 ΔAopyrG敲除菌株的构建
为了利用同源重组法获得米曲霉的ΔAopyrG敲除菌株(图1a),首先利用OE-PCR获得只含有AopyrG上下游序列的AopyrGLR片段(图1b),然后将其转化为野生型菌株的原生质体。转化混合物在含有5-FOA、尿苷和尿嘧啶的复苏培养基上培养,对长出的菌落提取DNA进行PCR验证。结果如图1c所示,用AopyrG的ORF引物进行PCR验证时发现,米曲霉野生型菌株可扩增出AopyrG的ORF片段,而转化菌株不能扩增出相应片段。用AopyrG的上下游引物AopyrG-P1和AopyrG-P2进行PCR扩增时发现,米曲霉野生型菌株扩增得到的片段比转化菌株扩增得到的片段长,以上结果说明米曲霉ΔAopyrG菌株构建成功。
2.2 ΔAoku70ΔAopyrG双敲除菌株的构建与验证
为了获得ΔAoku70ΔAopyrG双敲除菌株,先以Aoku70r-AfpyrG片段替换ΔAopyrG敲除菌中的Aoku70,然后利用5-FOA胁迫使得转化子基因组发生自身环化丢失AfpyrG实现Aoku70和AopyrG的双敲除(图2a)。用三对引物Aoku70-O1和Aoku70-O2(扩增Aoku70的ORF区)、Aoku70-L1和AfpyrG-O4(扩增L-AfpyrG区)、Aoku70-R2和AfpyrG-O3(扩增AfpyrG-R区)对Aoku70r-AfpyrG替换ΔAopyrG敲除菌中的Aoku70进行PCR验证。结果如图2b所示,野生型菌株只能扩增出Aoku70的ORF片段,而转化菌株只有Aoku70的ORF不能扩增出来。以上结果表明Aoku70r-AfpyrG成功替换了ΔAopyrG敲除菌的Aoku70基因。将上一步的转化子用5-FOA胁迫使得转化子基因组发生自身环化丢失AfpyrG,用AopyrG和Aoku70的ORF引物进行PCR验证。结果如图2c所示,野生型菌株可以同时扩增出AopyrG和Aoku70的ORF,而转化菌株都不能扩增出相应片段。但是,用引物Aoku70-P1和Aoku70-P2扩增Aoku70上下游跨越区时发现,野生型菌株扩增得到的片段比转化菌株扩增得到的片段长。综合以上结果表明ΔAoku70ΔAopyrG双敲菌株构建成功。
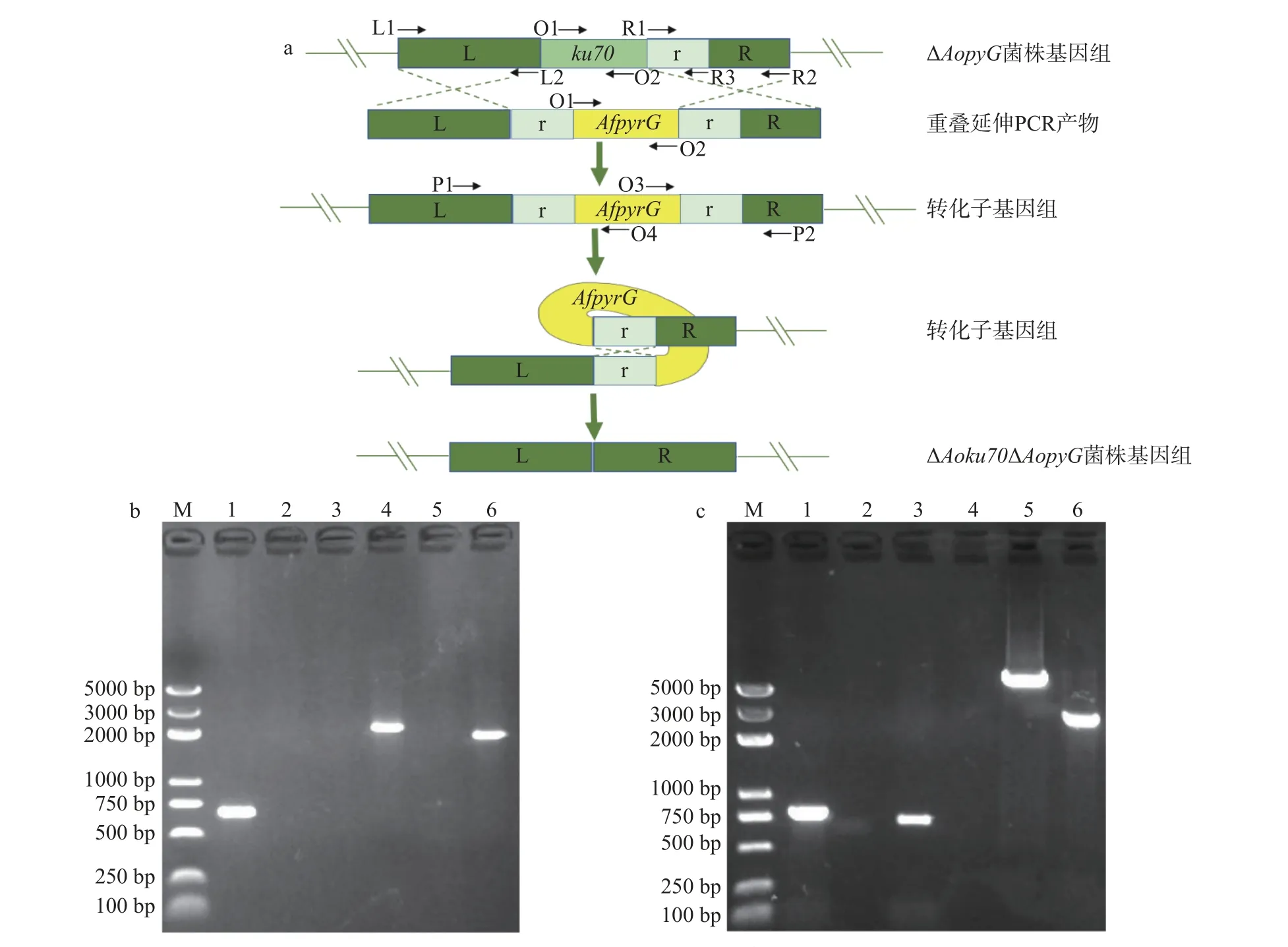
图 2 AopyrGAoku70基因敲除的验证Fig.2 Verification of the AopyrGAoku70 gene knockout
2.3 米曲霉核定位荧光菌株的构建
为了验证ΔAoku70ΔAopyrG双敲菌株的可用性,以该菌株为宿主利用同源重组技术将mCherry-AfpyrG片段插入到组蛋白H2B的ORF后面构建一个核定位荧光菌株(图3a)。用引物mCherry-O1和mCherry-O2对转化子的mCherry-ORF区进行PCR验证,用AoH2B上下游、mCherry内部和AfpyrG内部的引物对转化子进行AoH2B-mCherry区和mCherry-AfpyrG区的进一步验证。结果如图3b所示,三个片段大小均与预期相符,这说明AoH2B-mCherry荧光菌株构建成功。将验证成功的菌株孢子,经过萌发后,在激光共聚焦显微镜下观察其红色荧光的位置。结果如图3c所示,在细胞核中可以观察到红色荧光,进一步说明AoH2B-mCherry核定位荧光菌株构建成功。以上结果表明本研究构建的ΔAoku70ΔAopyrG双敲菌株可以用于米曲霉基因的改造研究。
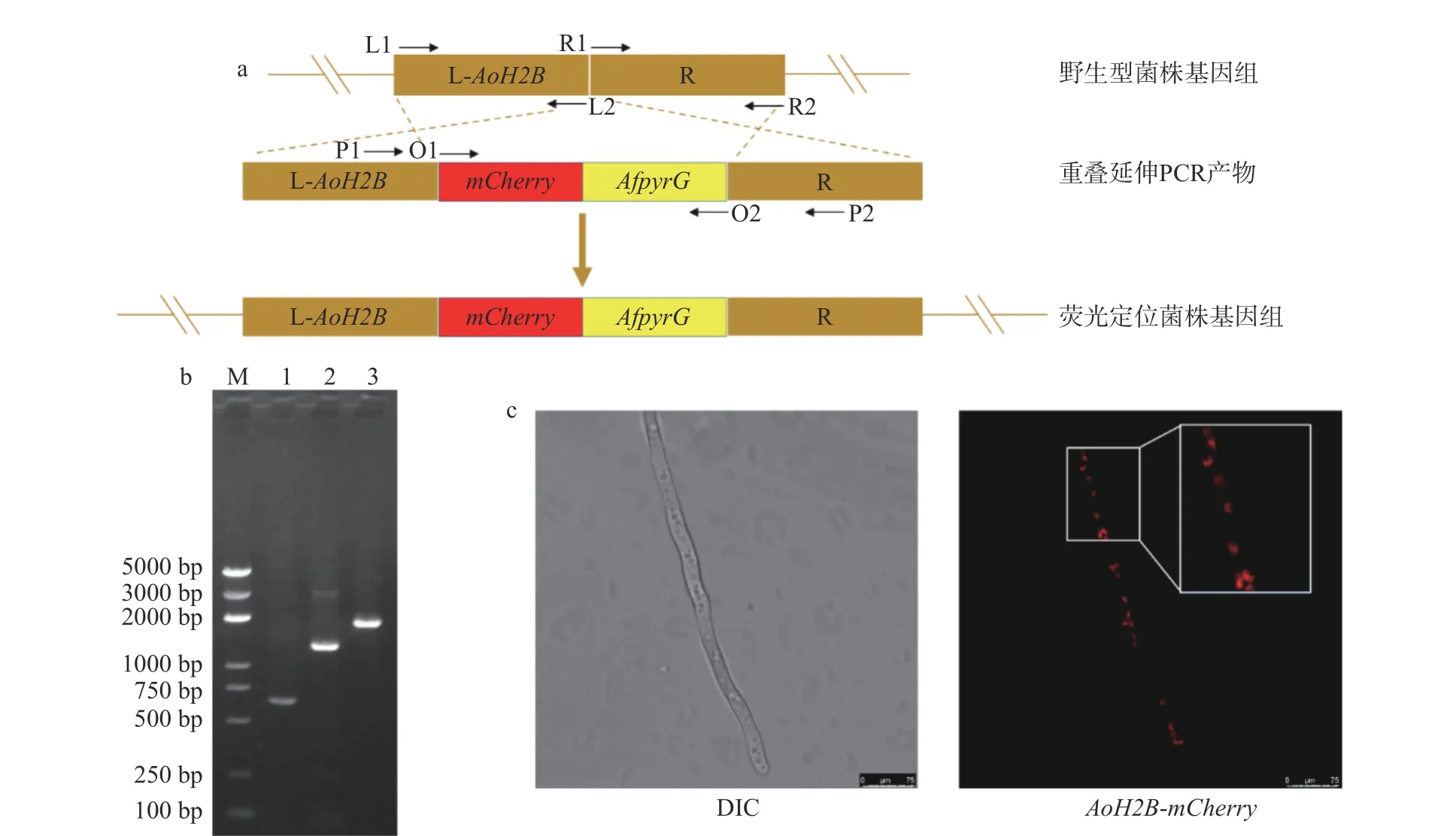
图 3 米曲霉核定位荧光菌株的构建Fig.3 Subcellular localization of AoH2B-mCherry in A. oryzae
2.4 米曲霉核定位荧光菌株的生长分析
为了研究插入mCherry基因对米曲霉生长的影响,分别将含有106个AoH2B-mCherry菌株和对照菌株(ΔAopyrGΔAoku70::AfpyrG)的孢子液接种到PDA培养基上,测量两种米曲霉在5 d内的生长直径。结果如图4所示,AoH2B-mCherry菌株的形态(图4a)和生长直径(图4b)与对照菌株相比没有明显差异(P>0.05),这表明mCherry基因的插入对米曲霉菌株的生长状况没有影响。后续研究可将融合了GFP荧光的目的基因转化至该核定位菌株,通过观察绿色和红色荧光的相对位置来确定目的基因的亚细胞定位,从而用于米曲霉基因的功能研究。
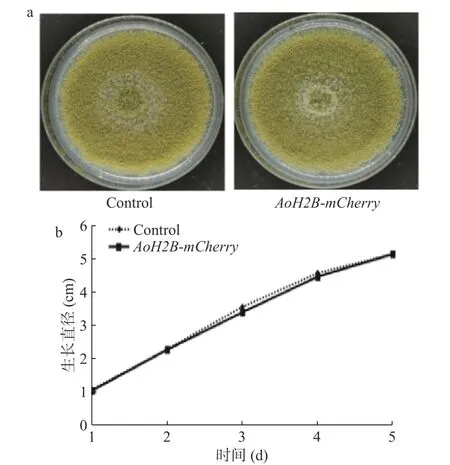
图 4 mCherry基因对AoH2B-mCherry菌株生长的影响Fig.4 The effect of mCherry gene on AoH2B-mCherry strain growth
3 讨论与结论
米曲霉的基因组序列虽已被测定,但是米曲霉的细胞壁成分特殊使其不能像酵母和大肠杆菌一样具有高的遗传转化效率[6,17],因此米曲霉遗传改良进展缓慢。建立高效的遗传转化体系首先需要考虑选择合适的遗传筛选标记。本研究通过敲除米曲霉中编码乳清酸核苷-5'-磷酸脱羧酶的pyrG基因,构建了以pyrG基因为筛选标记的遗传转化体系。与以往的主要以ble[18]、ptrA[19]、sdh[20-21]和hyg[22-23]等抗性基因作为筛选标记的遗传转化体系相比,以pyrG基因作为筛选标记不需要对抗性药物种类和浓度进行摸索,缩短了转化周期[24-26]。此外,pyrG基因的导入也不会使人们对食品安全产生疑虑,因此,基于pyrG基因进行遗传改良的米曲霉在生产应用中不会受到限制。
多数丝状真菌能利用非同源末端连接(Nonhomologous End Joining,NHEJ)途径修复DNA双链断裂(DNA Double Strand breaks,DSBs),使基因重组受阻[27-28]。NHEJ过程是由Ku70-Ku80异源二聚体、DNA依赖的蛋白激酶催化亚基和DNA连接酶IV-Xrcc4复合物所介导[29-30]。现有研究表明,在丝状真菌中敲除ku70基因可显著提高同源重组效率。Ninomiya等[29]在粗糙脉孢菌(Neurospora crassa)中敲除了ku70基因后发现,同源重组效率由野生型中的30%提高到了100%。Koh等[31]发现ku70缺失的圆红冬孢酵母(Rhodosporidium toruloides)突变体中,转化效率可达95.8%。Tu等[32]发现在ku70突变的亮盖灵芝(Ganoderma lucidum)中,CRISPR/Cas9基因编辑引起的靶基因插入效率能提高到96.3%,替换效率能提高到93.1%。本研究在ΔAopyrG单缺失的基础上,进一步敲除了Aoku70基因,获得了具有高效同源重组的营养缺陷型菌株ΔAoku70ΔAopyrG。
本研究以野生型的米曲霉RIB40为出发菌株,通过PEG-CaCl2介导的同源重组技术敲除了AopyrG和Aoku70两个基因,最终获得了尿苷/尿嘧啶合成和非同源末端连接途径同时缺陷的ΔAoku70ΔAopyrG突变体,并利用mCherry荧光定位验证了此遗传转化体系的可用性,为今后米曲霉RIB40的遗传改造和功能基因研究奠定了基础。
猜你喜欢
基层中医药(2022年4期)2022-07-22
四川蚕业(2022年1期)2022-06-06
太原理工大学学报(2021年6期)2021-11-25
汉字汉语研究(2021年2期)2021-08-30
海洋科学(2020年9期)2020-10-09
汉字汉语研究(2019年2期)2019-08-27
中国海洋大学学报(自然科学版)(2019年7期)2019-05-21
中国海洋大学学报(自然科学版)(2019年7期)2019-01-04
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
中国人兽共患病学报(2017年2期)2017-03-16
