
利伐沙班合成工艺改进*
2023-01-02 09:51刘玉键付新悦白万富
包头医学院学报 2022年12期
刘玉键 ,付新悦,周 微,白万富
(1.包头医学院药学院,内蒙古 包头 014040; 2.天士力控股集团有限公司研究院化学药品开发中心)
利伐沙班(Rivaroxaban Tablets),化学名为5-氯-氮-({(5S)-2-氧-3-[4-(3-氧-4-吗啉基)苯基]-1,3-唑烷-5- 基}甲基)-2-噻吩-羧酰胺,由德国拜耳公司研发。2008年在欧盟及加拿大等地上市,用于择期髋关节或膝关节置换手术成年患者,以预防静脉血栓形成。2009年6月在中国正式上市,商品名为拜瑞妥,2011年在美国上市,至今已在全球50多个国家上市[1]。利伐沙班作为新的口服抗凝药物,是一个具有高度选择性和竞争性直接抑制呈游离状态的Xa因子(FXa)的药物,对FXa的选择性是其他丝氨酸蛋白酶的10 000倍[2]。而且还可抑制结合状态的Xa因子以及凝血酶原活性,对血小板聚集没有直接作用。其抗凝作用依赖于通过内源性及外源性途径活化X因子为FXa,在凝血级联反应中发挥重要作用。因此,凝血酶的生成以及凝血酶原酶复合体的活性也会降低[3]。其具有生物利用度高,治疗疾病谱广,量效关系稳定,口服方便,出血风险低的特点。目前为止,利伐沙班主要用来治疗肺栓塞[4]、急性冠状动脉综合征急性期患者[5-6]、症状性急性室上性心动过速[7],以及非瓣膜性心房颤动患者心源性栓塞事件的预防[8]。利伐沙班的应用越来越多,单纯依靠进口已满足不了市场的需要。本文拟在利伐沙班原研路线的基础上,对其工艺路线进行优化改进,以满足产业化生产的要求。
1 材料
1.1仪器 DF-101S焦热式恒温加热磁力搅拌器(巩义市矛华仪器有限责任公司);ZF-7暗箱式三用紫外分析仪(上海嘉鹏科技有限公司);N-1210B旋转蒸发仪(东京理化器械株式会社);AUAMCE400核磁共振谱仪(美国BRUKER公司)。
1.2药品与试剂 N,N-羰基二咪唑(CDI)(分析纯,LI50P55),甲胺(分析纯,A1914072) ,均为上海阿拉丁生化科技股份有限公司;二氯甲烷(分析纯,20170525),无水乙醇(分析纯,20170509),四氢呋喃(分析纯,20170612),甲基磺酸(分析纯,20170123)、碳酸氢钠(分析纯,20170212),均为国药集团化学试剂有限公司。
2 实验方法与结果
在利伐沙班的原研合成路线基础上,在中和成盐、水解反应条件、粗品合成及精制等环节进行了优化,得到了优化后的利伐沙班合成路线。见图1。
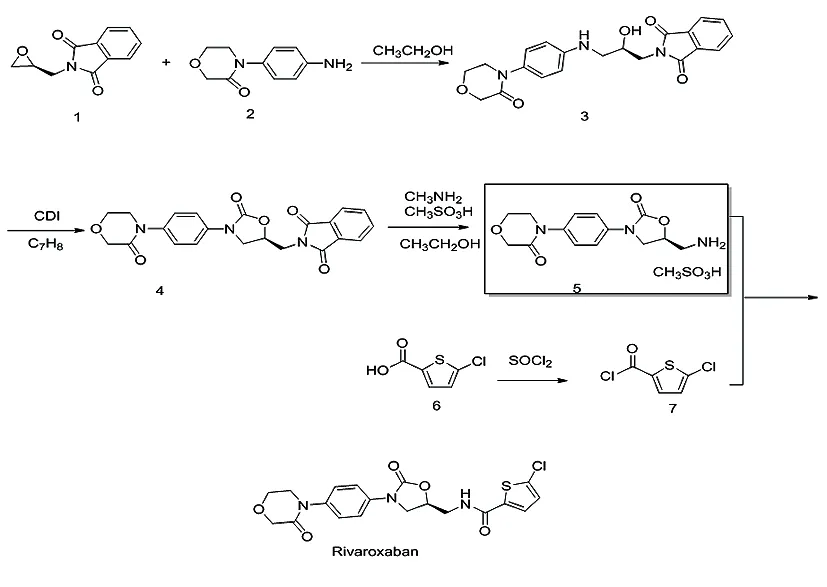
图1 利伐沙班优化后的合成路线
2.12-[(2R)-2-羟基-3-[[4-(3-氧代-4-吗啉基)苯基]氨基]丙基]-1H-异吲哚-1,3(2H)-二酮——化合物3的合成 向500 mL反应瓶内加入10 g(49.2 mmol)化合物1和9.45 g(49.2 mmol)化合物2,再加入100 mL无水乙醇和100 mL纯化水,缓慢升温至50-60 ℃,TLC跟踪监测反应至化合物2消失,反应约10 h结束。反应结束后将料液降至室温,搅拌析晶5 h。抽滤,滤饼用约25 mL无水乙醇淋洗后,干燥得17.74 g白色粉末,收率为91.2 %。1H-NMR(400 MHz,DMSO),δ:3.03(m,1H),3.16(m,1H),3.64(m,4H),3.91(t,2H),4.01(m,1H),4.14(s,2H),5.17(d,1H),5.66(t,1H),6.62(d,2H),7.03(d,2H),7.85(m,4H)。
2.22-[[(5S)-2-氧代-3-[4-(3-氧代-4-吗啉基)苯基]-5-噁唑烷基]甲基]-1H-异吲哚-1,3(2H)-二酮——化合物4的合成 向250 mL反应瓶内加入15 g(37.9 mmol)化合物3、7.5 g(46.3 mmol)CDI、114 mL甲苯,将料液升温,至回流后反应2 h,随后降温至室温,加入25 mL无水乙醇,于室温条件搅拌析晶2 h,过滤,滤饼用少量无水乙醇淋洗,干燥得到15.68 g白色粉末,收率98.2 %。1H-NMR(400MHz, DMSO),δ:3.71(t,2H),3.95(m,5H),4.22(t,3H),4.95(m,1H),7.41(d,2H),7.52(d,2H),7.89(m,4H)。
2.34-[4-[(5S)-5-(氨甲基)-2-羰基-3-唑烷基]苯基]-3-吗啉酮甲磺酸盐——化合物5的合成
2.3.1化合物4水解反应条件的优化 对于水解试剂的选择,本实验首先参考原研工艺使用甲胺作为水解试剂,经监测反应进程发现水解反应速度已较快,无需对其他水解试剂进行遴选考察,因此综合考虑物料成本等因素后确定采用甲胺水溶液作为水解试剂。随后对甲胺用量、反应温度和时间等对水解程度的影响进行了考察。结果表明,反应时间过短、甲胺用量少以及反应温度低等均会影响水解反应完全程度。因此经详细考察后最终确定甲胺与化合物4的摩尔比为6:1,水解温度控制在50~60 ℃,水解反应1 h,为最佳反应条件。见表1。

表1 不同反应条件对产品产率的影响
2.3.2成盐用酸的选择 本实验考察了其他的酸与中间体化合物4成盐对于产品的质量影响的情况。选择了甲酸、氢溴酸、硫酸和甲磺酸四种不同的酸进行中和成盐反应与浓盐酸进行比较,以便选择出更加适合产业化生产的酸。结果表明,用氢溴酸中和成盐会造成产品降解,效果最差,收率仅有65.56 %。用甲磺酸优化反应后收率与盐酸相当,而产品纯度要优于盐酸盐。因此最终确定使用该条件进行优化成盐。见表2。

表2 不同的酸对于产品产率的影响
2.3.3化合物5的合成 向250 mL反应瓶内加入15 g(35.6 mmol)化合物4、94.5 mL无水乙醇和15 mL甲胺水溶液,将料液升温至50~60 ℃;TLC跟踪监测反应终点,1 h反应结束后,将料液降至室温,用甲基磺酸调pH至3~4;继续搅拌析晶5 h;抽滤,滤饼用无水乙醇淋洗,干燥,得13.37 g白色粉末,收率97.0 %。1HNMR(400MHz,DMSO),δ:2.41(3H,s),3.24(t,2H),3.71(t,2H),3.85(m,1H),3.97(t,2H),4.22(t,3H),4.94(m,1H),7.43(d,2H),7.57(d,2H)。
2.4氯化、酰化反应——利伐沙班粗品制备
2.4.1利伐沙班粗品反应溶剂的优化 通过反应溶剂体系的对比考察表明,使用四氢呋喃-水反应体系产品收率高于丙酮-水体系。分别使用化合物5的盐酸盐、硫酸盐、甲磺酸盐和甲酸盐进行反应,考察结果表明盐酸盐和甲磺酸盐得到粗品的质量和收率相当;使用硫酸盐的收率次之,甲酸盐的收率最低。由此再次证明化合物5的制备工艺中可使用甲磺酸替代原研专利中的盐酸盐进行优化。见表3。

表3 酰化反应条件的优化
2.4.2利伐沙班粗品制备 向500 mL反应瓶中加入61 g(375.2 mmol)5-氯噻吩-2-羧酸,176 mL甲苯,滴加53 mL二氯亚砜,滴毕,回流反应3 h后降至室温25 ℃,得化合物7的甲苯溶液,直接用于酰化反应。向250 mL三颈瓶中加入12 g(30.9 mmol)中间体化合物5,搅拌溶解,向水层中加入101.8 mL四氢呋喃和8.5 g碳酸氢钠,开启降温,于0~15 ℃滴加上述化合物7的甲苯溶液。滴毕后将料液升温至50 ℃反应,TLC监测至化合物5消失。反应结束后,将料液降温至室温,搅拌析晶7.5 h,离心分离,滤饼用无水乙醇淋洗,甩干,干燥得 13.4 g利伐沙班粗品,收率98.74 %。1HNMR(400MHz,DMSO),δ:3.62(t,2H),3.71(t,2H),3.85(m,1H),3.97(t,2H),4.19(t,3H),4.84(m,1H),7.18(d,1H),7.41(d,2H),7.56(t,2H),7.69(d,1H),8.86(t,1H)。
2.5精制——利伐沙班成品制备
2.5.1利伐沙班精制过程中溶剂的选择 本步骤是对利伐沙班粗品的重结晶,各反应因素中溶剂对产品质量和收率影响最大,因此我们结合利伐沙班的溶解性、成品质量以及收率等因素对重结晶溶剂进行了重点筛选和考察。考察结果表明,按照专利[11]方法,使用乙酸进行重结晶(条件1)后,不仅成品收率较低,且因乙酸刺激性大、酸性强,对设备和人员的伤害均较强,不宜规模化生产使用。相对于乙醇-水体系,使用不同比例的丙酮-水体系重结晶(条件5、6、7)尽管得到的产品质量相差无几,但收率较低。乙醇-水体系中当二者体积比为2.5∶1时产品收率和质量均最佳(条件2),因此最终确定使用该溶剂体系作为利伐沙班粗品的精制溶剂。见表4。

表4 重结晶溶剂的选择
2.5.2利伐沙班成品制备 向250 mL反应瓶中加入4 g利伐沙班粗品,160 mL无水乙醇、20 mL纯化水、将料液升温至70~75 ℃,待固体全溶后加入活性炭,保温15~20 min,趁热将滤液压滤进入250 mL三颈瓶,将三颈瓶内料液降温至室温25 ℃,搅拌析晶8.5 h;离心分离,滤饼用无水乙醇淋洗,甩干,干燥6 h得3.87 g利伐沙班成品,收率96.86 %。
3 讨论
关于利伐沙班的合成,文献[9-10]报道中有很多方法,但是大多数存在合成原料成本高、操作复杂、合成路线过长等不足之处。其中拜耳公司申请的利伐沙班的制备专利[11]工艺操作简便、产品质量高,相对更适合产业化生产。该原研路线见图2。

图2 利伐沙班的原研合成路线
在上述利伐沙班原研路线[11]中,在由中间体化合物4水解成盐生成化合物5的过程中,收率仅为82.7 %,因此水解反应的试剂用量以及反应的各条件还需进一步优化改进。此外,拜耳公司对于其中的中间体化合物5的盐酸盐,即4-[4-((5S)-5-氨甲基-2-羰基-3-恶唑烷基)苯基]-3-吗啉酮盐酸盐,进行了专利保护。在专利文献[11]中,以丙酮-水体系参与利伐沙班粗品的合成步骤,收率较低;在粗品的精制的过程中,使用了具有酸性的乙酸进行重结晶,不适宜于进行产业化生产。
本文在以上路线的基础上,对其工艺路线进行了改进:(1)在2-[[(5S)-2-氧代-3-[4-(3-氧代-4-吗啉基)苯基]-5-噁唑烷基]甲基]-1H-异吲哚-1,3(2H)-二酮进行水解的时候,用甲磺酸代替盐酸进行中和成盐,从而避开了原研中的专利保护,生成了新的盐4-[4-[(5S)-5-(氨甲基)-2-羰基-3-唑烷基]苯基]-3-吗啉酮甲磺酸盐,同时对其水解条件进行了重新的调整;(2)在利伐沙班粗品的合成过程中,将酰化反应过程中丙酮-水体系换为四氢呋喃-水,即起始物料5-氯噻吩-2-羧酸与氯化亚砜发生氯代反应得到化合物7与经二氯甲烷萃取后的4-[4-[(5S)-5-(氨甲基)-2-羰基-3-唑烷基]苯基]-3-吗啉酮甲磺酸盐在四氢呋喃-水中发生酰化反应,得到利伐沙班粗品;(3)在利伐沙班粗品的精制过程中,将重结晶所用的溶剂乙酸换为乙醇-水,先将无水乙醇、纯化水和利伐沙班粗品升温至70~75 ℃,然后降温至室温20 ℃,搅拌析晶8.5 h,离心分离后,将滤饼干燥后,即可得到利伐沙班成品。
利伐沙班的合成工艺优化是以专利(11)中的合成方法为基础,进一步优化工艺路线,以便提高产品的质量,收率。在工艺优化过程中,以甲磺酸成盐的方式生成了利伐沙班的中间体4-[4-[(5S)-5-(氨甲基)-2-羰基-3-唑烷基]苯基]-3-吗啉酮甲磺酸盐,从而避免了专利保护。此外,通过探索,本实验发现与专利中的合成路线相比,最优的水解反应条件是甲胺与化合物4的摩尔比为6∶1,水解温度在50~60 ℃,水解反应1 h。最佳的酰化反应条件是化合物5的甲磺酸盐在四氢呋喃-水(v∶v =1∶1)中,于50~60 ℃条件下与化合物7反应1 h,得到利伐沙班粗品。以及在粗品的精制过程中,用乙醇-水体积比为2.5∶1代替冰乙酸时,不仅没有危害且产品收率和质量均最佳。与原研路线产品94.7 %的收率相比,经改进的合成路线总收率为96.86 %,提高了产品的收率。综上,改进后的合成路线不仅提高了产品的收率与纯度,而且操作更加绿色安全,对环境污染小,更加适合产业化生产。
猜你喜欢
发明与创新(2022年31期)2022-11-03
中国医药科学(2022年5期)2022-05-05
中国典型病例大全(2022年7期)2022-04-22
昆明医科大学学报(2020年11期)2020-12-28
工业设计(2020年6期)2020-07-30
中国生殖健康(2019年2期)2019-08-23
保健与生活(2019年6期)2019-07-31
中外医学研究(2019年13期)2019-07-11
上海医药(2016年11期)2016-06-30
分析化学(2014年6期)2014-07-10
