
散发性先天性心脏病相关SMAD1基因新突变研究
2022-12-31 10:34陈春英刘兴元杨奕清
国际心血管病杂志 2022年6期
陈春英 刘兴元 杨奕清
先天性心脏病是人类最常见的出生畸形,在活产新生儿中的发病率约为1%,可导致肺动脉高压、中枢神经系统发育延迟、脑损伤、心力衰竭、感染性心内膜炎以及致命性室性心律失常等严重并发症,甚至可致心源性猝死[1-2]。遗传学研究表明先天性心脏病主要是由遗传异常所致,除染色体异常和拷贝数变异外,目前已经发现了100 多个先天性心脏病致病基因,包括可导致家族性先天性心脏病的心脏转录因子基因SMAD1[3]。由于先天性心脏病具有显著的遗传异质性,且大多数先天性心脏病为散发[4-5],因此,有必要进一步研究SMAD1基因与散发性先天性心脏病的关系。
1 对象与方法
1.1 研究对象
2016 年3 月至2021 年10 月,入选202 例汉族散发性患儿为病例组,其中男性115 例,女性87 例,年龄为1~13 岁,平均年龄为(5±4)岁。对照组为238 名汉族健康志愿者,其中男性136 名,女性102 名,年龄为1~13 岁,平均年龄为(5±3)岁。所有研究对象均经过详细临床分析,包括家族史及病史回顾、体格检查和多普勒彩色心脏超声检查。根据阴性家族史、阳性心脏超声检查结果或心脏手术记录,诊断散发性先天性心脏病[6]。病例组和对照组均无先天性心脏病家族史,也均无可诱发先天性心脏病的环境危险因素。本研究遵循医学伦理学规范,并获得同济大学医学院附属同济医院伦理委员会的批准。经研究对象知情同意后,收集其临床资料和全血标本,用基因组DNA 提取试剂盒(德国Qiagen 公司)常规纯化基因组DNA。
1.2 方法
1.2.1SMAD1基因的扩增 通过聚合酶链反应(PCR)扩增SMAD1基因全部编码外显子、剪接位点以及部分5'和3'端非翻译区,引物DNA 序列见参考文献[3]。以每一研究对象的基因组DNA为模板,应用合成的上述特异性SMAD1扩增引物及热启动 DNA 聚合酶试剂盒(德国Qiagen 公司),在PCR 仪(美国Bio-Rad 公司)上对SMAD1基因片段进行扩增。PCR 混合物的总体积为50 μL,包括5×Q 溶液10 μL、双蒸水26.5 μL、dNTP(2.5 mmol/L)4 μL、上下游引物(20 μmol/L)各1 μL、10×PCR 缓冲液5 μL、基因组DNA(50 ng/μL)2 μL 和热启动DNA 聚合酶(5 U/μL)0.5 μL。PCR 的条件设置见参考文献[7]:95 ℃预变性15 min,随即进入38 个热循环,每个热循环包括94 ℃变性30 s、62℃ 退火30 s 和72 ℃延伸1 min,最后72 ℃延伸5 min。PCR 产物经1.5%琼脂糖凝胶电泳分离后割胶回收并用凝胶DNA 提取试剂盒(美国Bio-Tek 公司)进行纯化。
1.2.2SMAD1基因的Sanger 测序分析 以上述纯化的PCR 产物作为模板,使用1 条SMAD1基因扩增引物及DNA 循环测序试剂盒(美国Thermo Fisher Scientific 公 司)在PCR 仪(美 国Bio-Rad公司)上进行Sanger 测序反应。测序反应混合物的体积为20 μL,其中SMAD1基因DNA 片段(30 ng/μL)2 μL、预混合液8 μL、正向引物(2 μmol/L)2 μL、双蒸水8 μL。测序反应的条件[7]:30 个循环,每个热循环包括95 ℃变性20 s、50 ℃ 退火15 s 和60 ℃延伸l min。测序反应产物经纯化回收后在DNA 分析仪(美国Thermo Fisher Scientific 公司)上进行DNA 测序。将所测出的SMAD1基因序列与核苷酸数据库(https://www.ncbi.nlm.nih.gov/Nucleotide)中 的SMAD1基因序列(登陆号:NM_005900.3)进行比较分析,以发现SMAD1基因突变。如果识别出SMAD1基因突变,就测序分析238 名健康对照者的SMAD1基因,同时检索HGMD (http://www.hgmd.cf.ac.uk/ac/index.php)、SNP (https://www.ncbi.nlm.nih.gov/SNP)、PubMed (https://www.ncbi.nlm.nih.gov/PubMed)和万方数据库(https://g.wanfangdata.com.cn/index.html),以核实所发现的SMAD1基因突变是否是新突变。
1.2.3SMAD1基因突变的致病性模拟分析 通过在线计算机软件MutationTaster(http://www.mutationtaster.org/)预测分析SMAD1基因突变是否具有致病性。
1.2.4SMAD1基因突变体的功能研究 野生型SMAD1的表达载体SMAD1-pcDNA3.1 及其靶基因TBX20启动子驱动萤火虫荧光素酶(luc)表达的报告基因表达载体TBX20-luc 的构建可见参考文献[3]。以野生型SMAD1-pcDNA3.1 为模板,应用1 对长31 个碱基的互补引物(以突变点为中心)和定点诱变试剂盒(美国Stratagene 公司),通过PCR 获得突变型SMAD1-pcDNA3.1。应用DNA 酶Dpn I(美国NEB 公司)切除野生型SMAD1-pcDNA3.1模板并经过DNA 测序证实获得序列正确的突变型SMAD1-pcDNA3.1。COS7 细胞培养及多种表达载体共转染的方法见参考文献[3]。质粒转染后48 h收集、裂解COS7 细胞。用双报告基因(萤火虫荧光素酶和海肾荧光素酶表达基因)分析试剂盒(美国Promega 公司)在荧光分析仪(美国Promega公司)上定量分析细胞裂解液中荧光素酶的活性。以2 种荧光素酶的活性之比(萤火虫荧光素酶活性/海肾荧光素酶活性)表示靶基因TBX20启动子的转录活性[3]。
1.3 统计学分析
2 组连续变量如入选研究对象的年龄、靶基因TBX20启动子的转录活性等的比较用非配对Student'st检验;分类变量如入选研究对象的种族、性别等的比较用Pearson'sχ2检验或Fisher's 精确概率检验。以双侧检验概率值P<0.05 表示差异有统计学意义。
2 结果
2.1 鉴别出SMAD1基因新突变
本研究病例组(n=202)与对照组(n=238)入选者均为中国大陆汉族人,均无先天性心脏病家族史,两组间性别无统计学差异(经Pearson’sχ2检验P>0.05)和年龄(经Student’st检验P>0.05)也没有统计学差异。通过对202 例散发性先天性心脏病患儿的SMAD1基因进行Sanger 测序分析,在其中1 例3 岁的男性先天性右心室双流出道合并室间隔缺损患儿检测出1 种SMAD1基因杂合无义突变,即NM_005900.3: c.381T>A;p.(Cys127*)突变。该SMAD1基因突变不存在于238 名健康儿童,在HGMD、SNP、PubMed 和万方数据库也均无报道,表明该SMAD1基因突变是1 个新突变。该例先天性右心室双流出道合并室间隔缺损患儿的SMAD1基因c.381T>A 杂合突变及其纯合野生型对照DNA 序列见图1。
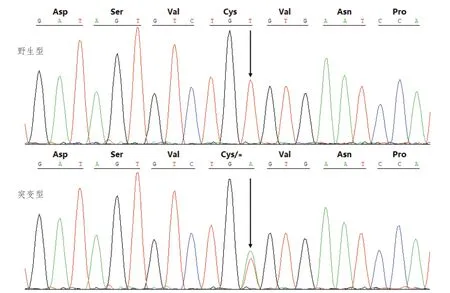
图1 SMAD1 基因的c.381T>A突变(杂合子)及其野生型(纯合子)对照DNA序列
2.2 SMAD1基因新突变c.381T>A有致病性
SMAD1基因新突变c.381T>A 被在线软件MutationTaster 预测为有致病性,这种预测的正确率约为1(>0.9999)。
2.3 Cys127*-突变型SMAD1对靶基因TBX20的转录激活功能丧失
在转染了多种基因表达载体的COS7 细胞,400 ng 的野生型SMAD1-pcDNA3.1 和等量(400 ng)的Cys127*-突 变 型SMAD1对靶基因TBX20启动子的转录激活效应分别约为11 倍(10.9328± 1.63)和1倍(1.14±0.41),2 组 之 间具有显著性差异(t=10.02,P=0.0006);而在同时转染了200 ng 的野生型SMAD1-pcDNA3.1 和等量(200 ng)的Cys127*-突 变 型SMAD1时,所 诱导的转录激活效应约为6 倍(5.80±0.71),显著低于400 ng 的野生型SMAD1-pcDNA3.1 所诱导的转录激活作用,2 组之间具有显著性差异(t=4.96,P=0.007)。
3 讨论
本研究在1 例散发性先天性心脏病(右心室双流出道合并室间隔缺损)患儿中发现了1 种新的SMAD1基因杂合无义突变,即NM_005900.3: c.381T>A;p.(Cys127*) 突变。该SMAD1基 因 突变不存在于238 名健康儿童,在HGMD、SNP、PubMed 和万方数据库也均无报道。在线计算机软件MutationTaster 模拟分析结果提示该突变具有致病性。报告基因分析表明Cys127*-突变型SMAD1对靶基因TBX20启动子的转录激活作用丧失,而TBX20功能障碍已经被证实可导致先天性心脏病[3]。因此,SMAD1基因新突变c.381T>A 或 p.(Cys127*) 很可能是该例散发性先天性心脏病(右心室双流出道合并室间隔缺损)患儿的分子病因。
人类SMAD1基因定位于4 号染色体4q31.21,编码一种属于SMAD 超家族的转录因子,由465 个 氨基酸残基组成[3]。SMAD1在整个胚胎发育期大量表达于心血管系统,可单独或与TBX20或MYOCD协同转录激活对心脏形态发育具有关键作用的靶基因(如TBX20、NKX2-5、TCTC1和MYH6)[8-10]。不仅如此,目前已经发现TBX20、NKX2-5、TCTC1、MYH6和MYOCD基因功能缺失性突变均可导致先天性心脏病[11-15]。本研究发现SMAD1基因功能缺失性新突变可导致右心室双流出道合并室间隔缺损。这些研究结果显示SMAD1基因单倍型不足是人类先天性心脏病的分子机制之一。
既往的实验动物研究发现,在胚胎发育期间,SMAD1可以在多种动物(包括小鼠、大鼠、蟾蜍和斑马鱼)的心血管系统大量表达,通过调节细胞生长、增殖、分化、凋亡,在心血管形态发育方面发挥重要作用[16-17]。在模型小鼠,Smad1基因敲除可导致胚胎死亡,主要原因在于胚胎不能着床于胎盘,胚胎中原始生殖细胞生成障碍及数量显著减少;而Smad1基因敲除杂合子小鼠发育无明显异常,这很可能与属于同一基因族的Smad5和Smad8基因的代偿作用有关,因为Smad5和Smad8基因的表达部位和蛋白功能特点与Smad1的相似[18-19]。进一步研究发现,尽管Smad1基因或Smad5基因单独敲除的杂合子小鼠发育正常,但Smad1基因和Smad5基因联合敲除的杂合子小鼠在胚胎期死亡,主要是因为小鼠胚胎心脏环化和偏侧化障碍[19]。此外,内皮细胞或平滑肌细胞特异性敲除Smad1基因小鼠可发生肺动脉高压、右心室肥厚和肺小动脉壁厚度增加[20]。这些实验动物研究结果支持SMAD1基因功能缺失性突变导致人类先天性心 脏病。
值得注意的是,SMAD1基因功能缺失性突变此前已经被发现可导致家族性动脉导管未闭、室间隔缺损和肺动脉狭窄[3]。本研究识别出SMAD1基因功能缺失性新突变可导致散发性右心室双流出道合并室间隔缺损,从而扩大了先天性心脏病相关SMAD1基因突变谱。
总之,本研究识别出了1 种新的SMAD1基因功能缺失性突变可导致右心室双流出道和室间隔缺损,这对先天性心脏病的早期精准预防有潜在的临床意义。
猜你喜欢
实用心脑肺血管病杂志(2022年8期)2022-12-06
世界最新医学信息文摘(2022年29期)2022-08-23
中国循环杂志(2022年5期)2022-06-02
临床医学工程(2022年5期)2022-05-19
天津医科大学学报(2021年4期)2021-08-21
中日友好医院学报(2021年1期)2021-04-14
广东蚕业(2021年1期)2021-03-18
心肺血管病杂志(2020年5期)2021-01-14
心肺血管病杂志(2020年5期)2021-01-14
山东医药(2020年9期)2020-05-20
