
AZD0914关键中间体(2R,6R)-2,6-二甲基吗啉的合成工艺研究
2022-12-30 03:09:24张银勇赵文豪杨玉社
合成化学 2022年12期
雷 响, 张银勇, 张 丹, 赵文豪, 孟 昕, 杨玉社,*
(1. 南京中医药大学 新中药学院,江苏 南京 210023; 2. 中国科学院 上海药物研究所,上海 201203)
细菌耐药性已成为全球公共卫生领域亟待解决的问题[1]之一,其中多药耐药性淋球菌(Neisseria gonorrheae)几乎无药可治[2]。目前,头孢曲松钠和头孢克肟作为医护人员治疗淋病的一线用药,同时,耐药的淋球菌也被发现[3-4]。因此,开发全新作用的抗淋球菌药物具有重大的临床意义[2-8]。
AZD0914(Zoliflodacin),化学名为(2R,4S,4aS)-11-氟-2,4-二甲基-8-[(4S)-4-甲基-2-氧代-1,3-恶唑烷-3-基]-1,2,4,4a-四氢-2′H,6H-螺[[1,2]-恶唑[4,5-g][1,4]恶嗪[4,3-a]喹啉-5,5′-吡啶]-2′,4′,6′(1′H,3′H)-三酮,是由AstraZeneca公司研制的一种新型螺嘧啶三酮类细菌拓扑异构酶II抑制剂,具有全新的结构和作用机制。AZD0914对敏感和耐药的淋球菌包括头孢曲松耐药的淋球菌均有较强的抗菌活性,现已进入临床III期研究阶段,主要用于单纯性淋病的治疗[2,9-14]。(2R,6R)-2,6-二甲基吗啉(1)是合成AZD0914的关键中间体之一。二甲基吗啉结构复杂,价格较贵,因此开发一条成本低、环境友好且适合产业化生产1的合成路线具有重要的现实意义。
文献报道了合成1的路线主要有2条。路线1(Scheme 1)[15]: (S)-2-(((三氟甲基)磺酰基)氧基)丙酸乙酯(2)与(R)-2-羟基丙酸乙酯(3)进行SN2反应得2,2′-氧基(2R,2′R)-二丙酸二乙酯(4),4再经四氢铝锂还原得到(2R,2′R)-2,2′-氧基双(丙烷-1-醇)(5),5随后与甲磺酰氯进行磺酰化得到(2R,2′R)-氧基双(丙烷-2,1-二基)二甲磺酸酯(6),6经过环合得到化合物(2R,6R)-4-苄基-2,6-二甲基吗啉(7),7最后经催化氢解制得化1。该路线中,市售原料2和3相对昂贵,且需要用到四氢铝锂等较为活泼危险的试剂,并且最后一步催化氢解用到的钯催化剂当量较大,此外每一步反应都需要柱层析纯化,无法保证最终产物的光学纯度。路线2为路线1的改进路线(Scheme 2)[16]。 (S)-1-(三苯甲氧基)丙-2-醇(8)与甲磺酰氯进行磺酰化得到(S)-1-(三苯甲基氧基)丙-2-基甲磺酸酯(9),9再与(R)-1-(三苯甲氧基)丙-2-醇(10)进行SN2反应得(((2R,2′R)-氧基双(丙烷-2,1-二酰基))双(氧基)双(甲烷四烷基)六苯(11),11经甲酸脱掉三苯甲基保护基得到5,后续反应同路线1。该路线中多数中间体为固体,便于纯化,且避免了昂贵危险试剂四氢铝锂的使用,成本较低。但在9与10的SN2反应中,9在碱性条件下极易发生消除反应生成大量的烯丙氧基三苯甲烷(IMP),使反应收率降低。
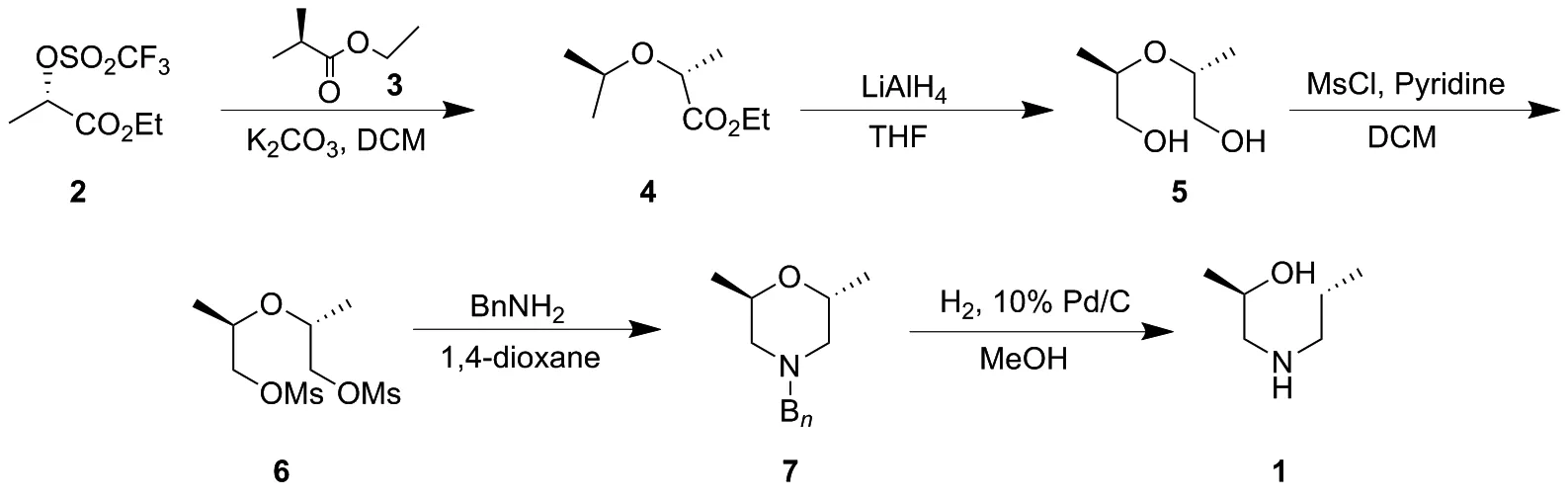
Scheme 1

Scheme 2
综合分析上述两条路线可知,路线2的大位阻中间体9与10的SN2反应收率极低,极大限制了该路线的进一步研究。因此本课题组在路线1的基础上进行重新设计和优化,如Scheme 3所示。路线1中,原料2售价高昂,因此需要研究2的合成工艺。原料3相对昂贵,而(R)-2-羟基丙酸甲酯(13)较便宜,因此选用13代替3可降低成本。在酯的还原中,采用硼氢化钠代替昂贵且危险的氢化铝锂。甲磺酸酯6为油状液体,不利于纯化,使用易于成固体的对甲苯磺酰氯进行磺酰化,有望得到固体(2R,2′R)-氧基双(丙烷-2,1-二基)双(4-甲基苯磺酸酯)(15)便于纯化,尤其是去除前述步骤中产生的立体异构体,从而保证1的光学纯度。之后通过设计成盐策略纯化7,使其以固体的形式析出。最后经过催化氢解可得到AZD0914关键中间体1。

Scheme 3
基于此,本文以廉价易得的(S)-2-羟基丙酸乙酯(12)和13为原料,将12与三氟甲磺酸酐反应得到2,随后2与13进行SN2反应得(R)-2-(((R)-1-甲氧基-1-氧代丙-2-基)氧基)丙酸乙酯(14),而14经硼氢化钠还原得到5,之后5与对甲苯磺酰氯反应得到15。用甲醇对15进行重结晶可除去立体异构体。最后15与16进行环化后再与对甲苯磺酸成盐得到(2R,6R)-4-苄基-2,6-二甲基吗啉对甲苯磺酸盐(16),产物经催化氢解、游离和蒸馏提纯制得AZD0914关键中间体1,总收率32%,纯度大于99%。
1 实验部分
1.1 仪器与试剂
Bruker AVANCE III 400/600 MHz型核磁共振仪。
三乙胺、吡啶、无水碳酸钾、硼氢化钠、1,4-二氧六环、甲醇。
1.2 合成
(1) (S)-2-(((三氟甲基)磺酰基)氧基)丙酸乙酯(2)的合成
温度为0 ℃条件下,在2 L三口瓶中将(S)-2-羟基丙酸乙酯(12)(100.000 g, 0.850 mol, 1.000 eq.)溶于超干DCM(1.000 L)。氩气保护下,滴加Tf2O(151.000 mL, 0.900 mol, 1.060 eq.),滴毕,搅拌20 min;缓慢滴加吡啶(73.000 mL, 0.900 mol, 1.060 eq.),溶液由无色澄清逐渐变为粉红色浑浊,滴毕,于0 ℃条件下反应,TLC监测(展开剂:乙酸乙酯 ∶石油醚=10 ∶1,V∶V,高锰酸钾显色)直至反应完全(约5 h)。反应完毕,加水淬灭反应,分液,用水(2×300 mL)洗涤有机相,水相用DCM(2×300 mL)萃取,合并有机相,依次用饱和食盐水(300 mL)洗涤,无水硫酸钠干燥、过滤,滤液浓缩得淡黄色液体2204.00 g,收率95%;1H NMR(400 MHz, CDCl3)δ: 5.22(q,J=7.0 Hz, 1H), 4.30(qd,J=7.1 Hz, 1.9 Hz, 2H), 1.71(d,J=7.0 Hz, 3H), 1.33(t,J=7.2 Hz, 3H);13C NMR(101 MHz, CDCl3)δ: 167.5, 123.3, 120.2, 117.0, 113.8, 80.3, 62.9, 18.2, 14.0; MS(ESI)m/z: 273.0{[M+Na]+}。
(2) (R)-2-(((R)-1-甲氧基-1-氧代丙-2-基)氧基)丙酸乙酯(14)的合成
将2(200.000 g, 0.799 mol, 1.000 eq.)溶于超干乙腈(2.000 L)中,加入(R)-2-羟基丙酸甲酯(13)(152.000 mL, 0.799 mol, 2.000 eq.)和无水碳酸钾(441.000 g, 3.200 mol, 4.000 eq.),在氩气保护的20 ℃条件下剧烈搅拌,用TLC监测(展开剂:乙酸乙酯 ∶石油醚=10 ∶1,V∶V,高锰酸钾显色)直至反应完全(12 h)。反应结束后,将反应液倒入铺有约0.500 cm厚硅藻土的布氏漏斗中过滤,滤饼用乙腈(3×300 mL)打浆后过滤,合并滤液、旋干。向所得粗产物中加入石油醚(400 mL)打浆后过滤,滤饼用PE(2×300 mL)打浆后过滤,合并滤液,用水(2×500 mL)洗涤滤液后再用饱和食盐水(500 mL)洗涤。合并水相,用石油醚(2×300 mL)反萃水相,合并有机相并用无水硫酸钠干燥,随后过滤、浓缩得淡黄色油状物14130.60 g,收率80%;1H NMR(400 MHz, CDCl3)δ: 4.25~4.14(m, 2H), 4.09(dq,J=10.2 Hz, 6.9 Hz, 2H), 3.74(s, 3H), 1.46(dd,J=6.9 Hz, 2.1 Hz, 6H), 1.28(t,J=7.1 Hz, 3H);13C NMR(151 MHz, CDCl3)δ: 173.6, 173.1, 74.2, 74.1, 61.0, 52.1, 18.9, 18.8, 14.3; (ESI)m/z: 227.2{[M+Na]+}。
(3) (2R,2′R)-2,2′-氧基双(丙烷-1-醇)(5)的合成
将14(130.000 g, 0.636 mol, 1.000 eq.)溶于超干四氢呋喃(1.300 L)中,加入甲醇(53.000 mL, 1.270 mol, 2.000 eq.),升温至50 ℃,分批加入硼氢化钠(48.000 g, 1.270 mol, 2.000 eq.),待体系平稳后升温至回流,用TLC监测(展开剂:乙酸乙酯 ∶石油醚=1 ∶5,V∶V,高锰酸钾显色)直至反应完全(约5 h)。反应结束后冷却至室温,在冰浴条件下,加入浓盐酸调pH=3~4,析出大量固体。搅拌反应1 h后过滤,用四氢呋喃(500 mL)冲洗滤饼,然后向滤液中加入碳酸钠(135.000 g, 1.270 mol, 2.000 eq)调节pH=7,随后过滤,滤液浓缩干,加入二氯甲烷(500 mL)打浆,过滤后滤液用无水硫酸钠干燥,过滤,浓缩干后得淡黄色油状物570.30 g,收率83%;1H NMR(400 MHz, CDCl3)δ: 3.78~3.62(m, 2H), 3.56(dd,J=11.4 Hz, 3.4 Hz, 2H), 3.46(dd,J=11.4 Hz, 7.8 Hz, 2H), 2.21(s, 2H), 1.12(d,J=6.2 Hz, 6H);13C NMR(101 MHz, CDCl3)δ: 73.2, 66.6, 16.3; MS(ESI)m/z: 157.2{[M+Na]+}。
(4) (2R,2′R)-氧基双(丙烷-2,1-二基)双(4-甲基苯磺酸酯)(15)的合成
将5(70.000 g, 0.522 mol, 1.000 eq)溶于超干DCM(700 mL)中,0 ℃条件下加入TsCl(218.800 g, 1.148 mol, 2.200 eq.)、三乙胺(218.000 mL, 1.565 mol, 3.000 eq.)和DMAP(6.350 g, 0.052 mol, 0.100 eq.),然后升至室温反应3 h,待TLC监测(展开剂:乙酸乙酯 ∶石油醚=2 ∶1,V∶V,碘显色)反应完后,加水(700 mL)淬灭反应,分液,水相用DCM(3×300 mL)萃取,合并有机相,先后用2N HCl水溶液(3×300 mL)和饱和食盐水(300 mL)洗涤有机相,最后经无水硫酸钠干燥后过滤、浓缩,得到棕色固体粗品211.0 g,将所得固体用甲醇(2×300 mL)重结晶,过滤得到的滤饼用甲醇(50 mL)淋洗,烘干得白色固体15139.00 g,收率67%, m.p.75~78 ℃,纯度99.7%[Agilent 1100系列LC系统(PLATISILODS 5.0 μm 250.0 mm×4.6 mm)测定,流动相为水-乙腈(60 ∶ 40,V∶V),检测波长为254 nm,柱温30 ℃,流速1.0 mL/min],ee99.99%,de99.8%[HPLC法:CHIRALPAK IA(4.6 mm×250.0 mm, 5.0 μm),流动相为正己烷-乙醇(90 ∶10,V∶V),检测波长为254 nm,柱温25 ℃,流速1.0 mL/min];1H NMR(400 MHz, DMSO-d6)δ: 7.84~7.68(m, 4H), 7.47(d,J=8.0 Hz, 4H), 3.92(dd,J=10.4 Hz, 3.3 Hz, 2H), 3.78(dd,J=10.4 Hz, 6.2 Hz, 2H), 3.66(td,J=6.3 Hz, 3.3 Hz, 2H), 2.41(s, 6H), 0.89(d,J=6.3 Hz, 6H);13C NMR(101 MHz, DMSO)δ: 144.9, 132.3, 130.1, 127.6, 73.1, 71.3, 21.1, 16.5; MS(ESI)m/z: 443.2{[M+Na]+}。
(5) (2R,6R)-4-苄基-2,6-二甲基吗啉对甲苯磺酸盐(16)的合成
将15(80.000 g, 0.180 mol, 1.000 eq.)溶于1,4-二氧六环(800 mL)中,加入苄胺(86.800 g, 0.810 mol, 4.500 eq.),并在100 ℃条件下进行反应,用TLC监测(展开剂:乙酸乙酯 ∶石油醚=1 ∶5,V∶V)直至反应完全(约12 h)。然后将反应体系降至室温,加入TsOH·H2O(54.800 g, 0.290 mol, 1.600 eq.),搅拌反应2 h,慢慢析出大量固体,随后过滤,滤饼用1,4-二氧六环(400 mL)冲洗,滤液浓缩干,加入乙酸乙酯(800 mL)和饱和碳酸氢钠溶液(200 mL),分液后有机相先后用饱和氯化铵(2×200 mL)和饱和食盐水(200 mL)洗涤,无水硫酸钠干燥后过滤。向滤液中加入TsOH·H2O(34.200 g, 0.180 mol, 1.000 eq.),搅拌反应30 min,析出大量固体,过滤,滤饼用乙酸乙酯(20 mL)淋洗后烘干,得到乳白色固体1666.46 g,收率96%, m.p.162~167 ℃;1H NMR(400 MHz, CDCl3)δ: 11.19(s, 1H), 7.84~7.77(m, 2H), 7.55~7.48(m, 2H), 7.42(qd,J=8.7 Hz, 7.8 Hz, 3.6 Hz, 3H), 7.19(s, 3H), 4.66(dd,J=13.0 Hz, 3.3 Hz, 1H), 4.40~4.17(m, 2H), 4.07(dd,J=13.0 Hz, 5.9 Hz, 1H), 3.72(dd,J=11.8 Hz, 2.3 Hz, 1H), 3.02(d,J=12.2 Hz, 1H), 2.83(td,J=12.0 Hz, 11.3 Hz, 4.8 Hz, 1H), 2.37(s, 7H), 1.51(d,J=7.2 Hz, 3H), 1.14(d,J=6.2 Hz, 6H);13C NMR(151 MHz, CDCl3)δ: 142.2, 140.3, 131.8, 130.4, 129.5, 129.0, 127.5, 126.1, 67.0, 61.9, 61.8, 57.3, 52.7, 21.5, 18.7, 16.7; MS(ESI)m/z:128。
(6) (2R,6R)-2,6-二甲基吗啉(1)的合成
将16(66.000 g, 0.175 mol, 1.000 eq.)溶于甲醇(530 mL)中,加入Pd(OH)2/C(1.320g, 含钯20%,含水约50%,质量分数2%),在50 ℃条件下催化氢解,用TLC监测(展开剂:乙酸乙酯 ∶石油醚=1 ∶3,V∶V)直至反应完全(约7 h)。随后过滤,滤液浓缩得油状物,并将其溶于水(150 mL)中,冰浴下加入氢氧化钠(8.500 g, 0.210 mol, 1.200 eq.)后再加入氯化钠固体至出现不溶现象。加入二氯甲烷(4×150 mL)萃取,合并有机相,用无水硫酸钠干燥、过滤,滤液浓缩(200 hPa以上,40 ℃),减压蒸馏纯化(水泵,外温80~120 ℃,温度计75 ℃出产物)得到无色透明液体117.40 g,收率86%,含量102%;1H NMR(400 MHz, CDCl3)δ: 4.02~3.76(m, 2H), 2.91(dd,J=12.3 Hz, 3.4 Hz, 2H), 2.52(dd,J=12.2 Hz, 5.8 Hz, 2H), 1.18(d,J=6.5 Hz, 6H);13C NMR(151 MHz, CDCl3)δ: 66.6, 51.2, 17.7; MS(ESI)m/z:116.1{[M+H]+}。
2 结果与讨论
2.1 合成工艺研究
(1)2的合成
首先考察了碱的种类及用量对反应收率和时间的影响,其结果如表1所示。由表1可知,以2,6-二甲基吡啶或吡啶为碱时2的收率相对较高(Entry 1, Entry 4),然而2,6-二甲基吡啶的价格较昂贵且后处理中不易除掉,因此选择吡啶作为碱对反应进行进一步优化。当以吡啶为碱时,投料顺序对反应有很大影响。加入三氟甲磺酸酐后再滴加吡啶,产率相对较高(表2)。最终确定反应以吡啶(1.060 eq.)作为碱,温度设置为0 ℃,先加入三氟甲磺酸酐(1.060 eq.),之后缓慢滴加吡啶。
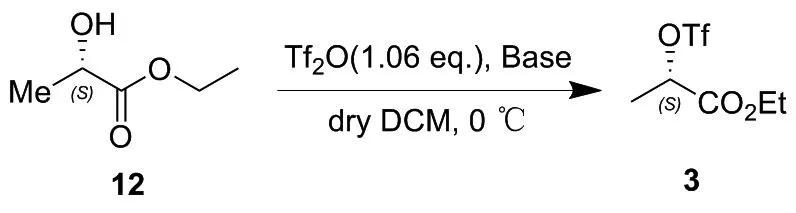

表1 碱对反应的影响Table 1 Effect of base on the reaction

表2 加料顺序对反应的影响Table 2 Effect of loading sequence on the reaction
(2)14的合成
合成14时,最初使用R-乳酸乙酯和2进行SN2反应,后续研究中将其替换成更廉价的R-乳酸甲酯。此外,原路线采用的1,2-二氯乙烷溶剂属于毒性较强的一类溶剂,不适合大规模生产使用。因此,本文对反应条件进行了优化,其结果如表3所示。由表3可以看出,使用溶解性更好的溶剂(Entries 6~9),反应收率会大大降低或者得不到产物。而当碳酸钾的当量降低后,反应时间将延长(Entry 11)。因此,最终确定以R-乳酸甲酯为原料,4.0 eq. K2CO3为碱,乙腈为溶剂,在25 ℃条件下进行反应(Entry 2)。

表 3 化合物14合成条件的优化Table 3 Optimal conditions for synthesis of 14
(3)5的合成
在5的合成中最初采用的是四氢铝锂作还原剂,该试剂价格昂贵且危险,后处理方式不利于放大生产,因此本文对反应中还原剂的选择进行了考察。表4分别考察了硼氢化钠/氯化锌体系以及硼氢化钠/甲醇体系将酯还原为醇的效果。由表4(Entry 2)可以看出,在25 ℃条件下的硼氢化钠/氯化锌体系中,以THF作为溶剂反应22 h后原料仍有大量剩余,说明该反应体系不能较好地对14进行还原。接着采用硼氢化钠/甲醇体系对14进行还原,从表4(Entry 3)可以看出,在50 ℃条件下以THF为溶剂反应5 h后,反应效果较好。因此,本文以硼氢化钠/甲醇体系为还原体系,并进一步对硼氢化钠的用量进行考察,其结果如表5所示。由表5可以看出,硼氢化钠用量较大时,5的收率反而相对较低(表5, Entry 1~2),这可能与硼氢化钠的副产物硼酸固体会包裹产物有关。由表5(Entry 4)可知,当硼氢化钠用量为2.00 eq.时,既可节约成本,收率也较高,因而本文以加量为2.000 eq.硼氢化钠作为还原剂,在甲醇的存在下将酯还原为醇。而从后处理的过程中可以发现,产物极易溶于水且不利于萃取,因此,采取调节pH产生固体,过滤、浓缩的方式来完成后处理。另一方面,在反应过程中会有气泡生成,因而通过采取分批加入硼氢化钠的方式使反应能够平稳进行。在调节pH过程中,用HCl的甲醇溶液淬灭,使硼烷生成容易旋蒸除去的硼酸甲酯(m.p. 68 ℃),但其与浓盐酸淬灭效果相当,因而最终选择浓盐酸调节pH。

表4 还原剂种类对反应的影响Table 4 Effect of reducing agent type on reaction
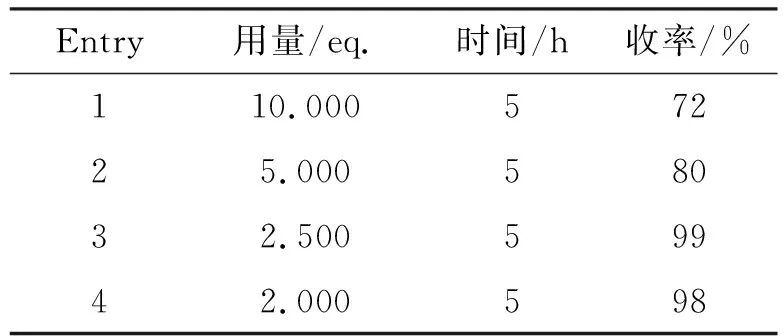
表5 NaBH4用量对反应的影响Table 5 Influence of dosage of NaBH4 on reaction
(4)15的合成
在15的合成中最初采用的是甲磺酰氯作为磺酰化试剂,但所得产物为油状物,很难通过工艺手段对其进行纯化。因此尝试使用对甲苯磺酰氯作为磺酰化试剂,然后加入催化量的DMAP可使反应时间缩短至3 h,所得产物为固体。随后,对产物纯化方法进行研究(表6),最终选择甲醇作为重结晶溶剂(Entry 5),可显著提高15的纯度。

表6 化合物15纯化条件分析Table 6 Analysis of the purification conditions of 15
(5)16的合成
苄胺在此反应中既作为碱也作为环化试剂,因此该步工艺难点是除掉反应中多余的苄胺。本文在前期优化的过程中试图降低苄胺的当量(表7),然而发现苄胺当量过少时反应不彻底,因此经综合考虑最终确定苄胺用量为4.000 eq.(Entry 3)。通过加入TsOH·H2O与苄胺形成苄胺盐,过滤除掉,滤液经萃取后加入TsOH·H2O,有固体析出,过滤后可得产物。

表7 BnNH2当量分析Table 7 Analyisis of BnNH2 equivalent
(6)1的合成
该步反应优化了溶剂、温度及Pd(OH)2/C用量(质量分数)。由表8可知,将Pd(OH)2/C用量降至2%时(Entry 6),原料依然可以催化氢解彻底,因此选定反应条件为:以甲醇为溶剂,温度为50 ℃, Pd(OH)2/C用量为2%。

表8 Pd(OH)2/C用量分析Table 8 Investigation of Pd(OH)2/C usage
2.2 化合物15的绝对构型
化合物15(M=442.53 g/mol)为单斜晶系,属于C2(no.5)空间群,晶胞参数为a=25.0619(14) Å,b=5.5811(3) Å,c=7.7350(4) Å,α=90.000°,β=92.846(2)°,γ=90.000°,V=1080.58(10) Å3,Z=2,T=170.0 K,μ(MoKα)=0.285 mm-1,wR2=0.0652。

图1 15的单晶结构Figure 1 Single crystal structure of 15
本研究对AZD0914关键中间体(2R,6R)-2,6-二甲基吗啉(1)的合成路线进行了优化。在二酯还原步骤中用较温和的硼氢化钠代替危险的氢化铝锂,将所得到的二醇与对甲苯磺酰氯反应得到的中间体为固体,其可通过重结晶除去杂质和异构体。而在成环氨化反应中通过加入对甲苯磺酸可方便除去剩余的苄胺。优化后的工艺反应条件温和,无需柱层析,所得目标产物1异构体含量低、纯度高(总收率32%,产品纯度99%以上),操作简洁,为工业化生产提供了新的选择。
猜你喜欢
昆明医科大学学报(2021年2期)2021-03-29 07:42:14
上海计量测试(2020年1期)2020-03-18 02:31:22
铜仁学院学报(2018年6期)2018-07-05 09:47:36
环境与发展(2018年3期)2018-05-10 11:21:48
湖南理工学院学报(自然科学版)(2017年4期)2018-01-25 06:01:08
无机盐工业(2017年5期)2017-05-25 00:37:34
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
中国洗涤用品工业(2016年2期)2016-02-28 19:03:18
环境科技(2015年1期)2015-11-08 12:10:50
中国塑料(2015年2期)2015-10-14 05:34:31
