
儿童囊性纤维化并巨结肠同源病1例
2022-12-17 06:10徐宇鹏姜斌黄磊
南京医科大学学报(自然科学版) 2022年12期
徐宇鹏,姜斌,黄磊
南京医科大学附属儿童医院普外科,江苏 南京 210019
囊性纤维化(cystic fibrosis,CF)是由位于染色体带7q31.2 上囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)基因突变引起的一种常染色体隐性遗传病,主要表现为慢性呼吸道疾病、慢性鼻窦炎、胰腺炎、营养不良和男性不孕等[1-3]。巨结肠同源病(Hirschsprung’s allied disease,HAD)是有类似先天性巨结肠(Hirschsprung’s disease,HD)临床表现,如腹胀、肠管扩张、慢性便秘,直肠病理检查存在肠神经节细胞的疾病[4]。囊性纤维化在我国极为罕见,白人群体发病率约为1/3 000[1],现报道1例CF患儿合并HAD的临床资料,用于提高对该疾病的认识。
1 病例资料
患儿,女,2 岁9 个月,因“反复咳喘1 年余”入院。入院体格检查:神志清,精神可,呼吸平稳,可及轻度吸凹征,双肺呼吸音粗,可闻及喘鸣音,心腹查体未见明显异常,未见杵状指。入院时实验室检查:丙氨酸氨基转移酶(ALT)87.0 U/L,天门冬氨酸氨基转移酶(AST)56.0 U/L,肌酸酶同工酶32.0 U/L,白蛋白39.7 g/L,过敏原IgE 示粉尘螨强阳性,痰培养检查示铜绿假单胞菌多发耐药。入院时胸部CT(图1A)示两肺炎症,小气道病变伴感染。既往史:患儿新生儿期因肠梗阻于当地医院行造瘘手术,10月龄行关瘘手术,术后至本次住院治疗前无再次肠梗阻住院治疗史,因呼吸道感染反复外院治疗史。父母体健,有一14岁哥哥,有鼻炎病史。
患儿入院后于呼吸科治疗,入院5 d 患儿出现腹痛、呕吐,伴脐周压痛,血常规示:白细胞计数18.39×109个/L,中性粒细胞百分比65.7%,腹部B 超示腹腔胀气,入院8 d 腹痛较前稍缓解,腹稍膨,触诊软,有排气,停止排便2 d,入院9 d出现腹胀明显,肠鸣音减弱,停止排气排便,无明显腹膜炎表现,腹部CT(图1B、C)示:结肠窄,部分结肠壁增厚,小肠增宽,内容物多;肝脏密度普遍性减低;两肾密度不对等,右侧偏高,右肾上盏积水;两肺炎症。腹部立位片(图1D)可见部分肠管积气并液平。普外科会诊后行剖腹探查。术中见肠管粘连明显,见距回盲部60 cm左右回肠大便填充,肠管僵硬水肿,结肠至直肠萎瘪,根据冯杰雄等[4]HAD 病理检查对小肠和乙状结肠肠壁进行全层活检的建议,取直肠及乙状结肠交界处黏膜活检,快速病理示可见成熟神经节细胞,取距回盲部25 cm原关瘘处肠管切开减压,见黏液样粪便排出,放置阑尾支架管,送阑尾及关瘘处肠管病理检查。术后病理示:阑尾结构存在,腔内积粪,局部黏膜及黏膜下层坏死,各层见大量炎症细胞浸润,以中性粒细胞为主;阑尾系膜亦见大量炎症细胞浸润;阑尾肌间神经丛内神经节细胞,大部分发育较差(图2A)。肠管黏膜下神经丛及肌间神经丛内找到神经节细胞,大部分发育较差(图2B)。免疫组化结果:黏膜下神经丛及肌间神经丛CR(+),S-100(+),Syn(+),PGP9.5(+)。符合HAD病理特征。
术后患儿出现反复咳嗽、咳痰,腹胀较缓解,痰培养检查示屎肠球菌阳性,复查血常规基本正常,复查胸片示两肺炎症,右上肺实变逐渐吸收。肝功能指标可降至正常,入院21 d 复查肝功能示:ALT43.0 U/L,AST 56.0 U/L。入院27 d 复查胸腹部立位片示(图1E):两肺肺炎较前好转,不全性肠梗阻需考虑。入院30 d好转出院。出院7 d复查腹部CT提示结肠内容物较多等(图1F、G)。术后2 个月随访患儿营养不良,体重增长缓慢,无其他明显不适。
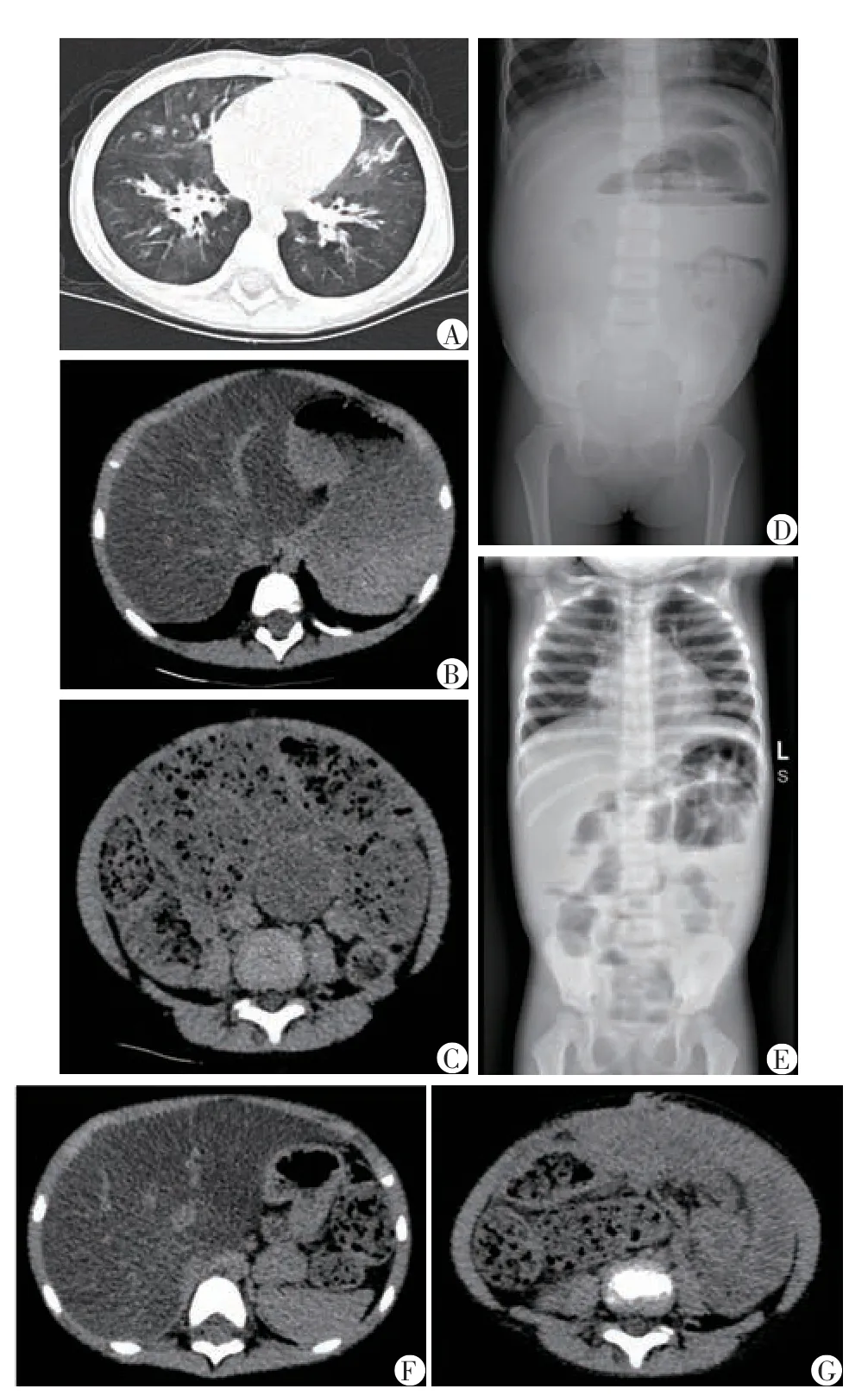
图1 患儿影像学资料
病程中与患儿家属沟通并取得知情同意后完善全外显子检查,基因测序由北京全谱医学检验实验室完成。结果显示,患儿CFTR 基因存在复合杂合变异(图3),分别为来自母亲的杂合突变:第3 外显子第223 号核苷酸由胞嘧啶变为胸腺嘧啶,导致第75 位氨基酸由精氨酸变为终止密码子(c.223C>T,p.Arg75X);来自父亲的杂合突变:第12外显子第1 624号核苷酸鸟嘌呤变为胸腺嘧啶,导致第542位氨基酸由甘氨酸变为终止密码子(c.1624G >T,p.Gly542X)。根据基因检测结果及病理结果,患儿最终诊断为CF合并HAD。

图2 患儿病理学资料(×200)
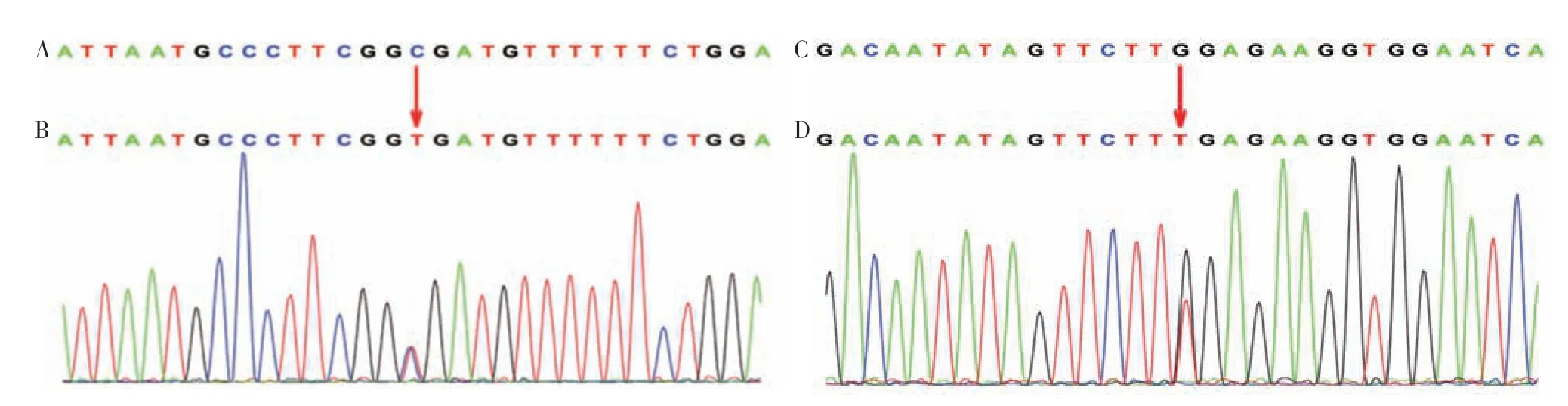
图3 患儿基因检测结果
2 讨论
CFTR 是一种主要转运氯离子和碳酸氢盐的cAMP依赖性阴离子通道,存在于气道、胰腺、肝胆、肠道、生殖系统、汗腺等多种上皮细胞顶膜中,起调节水、盐分泌、吸收和维持酸碱平衡的作用,其功能异常使导管上皮细胞水分、Cl-和HCO3-分泌减少,导致黏液黏稠,产生多系统症状[1-3]。
大多数CF患者出生后不久出现症状,最常见的临床表现是呼吸道感染和营养不良,呼吸衰竭是CF患者主要的死亡原因。CF 患者常因呼吸道感染反复住院治疗,给与积极营养支持、清除气道分泌物、抗感染治疗可明显减轻患者症状,延长患者生存时间,提高生存率[1,5]。囊性纤维化相关肝病(cystic fibrosis-related liver disease,CFLD)是CF 患者次要死亡原因,CFTR 在肝内和肝外胆管细胞中表达,不表达于肝细胞中,CFTR 功能障碍导致胆汁黏稠、胆管阻塞、胆汁酸潴留,临床表现为肝大、静脉曲张出血、肝酶持续升高和微胆囊形成,后期发展为肝硬化[1,6]。远端肠梗阻综合征(Distal intestinal obstruction syndrome,DIOS)是CF患者常见的肠道并发症,主要见于胰腺功能不全、胎粪性肠梗阻、剖腹手术和肺移植的患者,特征是黏性粪便阻塞末端回肠,典型临床表现为间歇性腹部绞痛或急性腹痛,可局限于右下腹,伴有呕吐、腹胀,右侧髂窝内可触及回盲部囊性肿块,典型腹部平片为全腹部小肠扩张,右侧髂窝有“泡状”肿块[7]。部分CF 患者表现有反复腹泻、便秘、脂肪泻和营养不良,通常认为是由胰腺外分泌功能不全引起脂肪、蛋白质和糖类吸收障碍所导致[8]。CFTR表达于肌间神经丛上,可调节肠道胆碱能神经传递,CF患者慢性便秘可能与其表达异常有关,对CF 患者末端回肠的病理检查显示,肠神经元数量无明显改变[9-10]。文献复习时发现CF患者伴有肠神经元性发育异常的病例报道非常罕见,Wildhaber等[11]报道1 例合并肠神经元发育不良的CF患儿,伴有典型DIOS表现,其术后病理显示全结肠并延伸至小肠的典型的B 型肠神经元发育不良的组织学表现,该报道与本例较为相似。
根据2017年囊性纤维化基金会提出的最新CF诊断标准共识指南[3],CF的诊断以汗液实验中氯离子浓度≥60 mmol/L 或具有2 个CFTR 基因突变或2 个CFTR 生理实验(肠电流测量实验和鼻电位差实验)阳性为主。
本例患儿下呼吸道感染反复发作,与CF患者常见临床症状相符,予对症治疗均可改善,肺部CT 未提示支气管扩张表现,考虑为长期呼吸系统治疗减缓肺部症状发展。入院后予还原型谷胱甘肽、复方甘草酸苷保肝治疗后肝酶指标可降至正常,后复高考虑为长期静脉营养的不良反应。CT 提示肝脏密度普遍性减低,考虑患儿伴有CFLD,肝胆系统症状以肝功能异常、肝影像学异常为主,无明显黄疸、呕血、肝大、肝区疼痛等肝病临床症状,考虑为患儿年龄较小,肝功能损害较轻。CFLD 治疗上目前争议较大,对于无明显临床症状的CF 患者,仍需要长期检测肝酶指标、肝脏影像学表现,给予长期保肝支持。患儿术中回盲部可见大量粪便充填,肠切开减压可见黏液样粪便排出,术后患儿反复不全性肠梗阻表现,结合既往剖腹手术史,考虑伴有DIOS。术中发现结肠肠管僵硬水肿、充盈不良,术后病理检查符合HAD 病理特征,结合目前研究未发现CFTR功能障碍直接影响肠神经元发育的证据,考虑本例患儿合并有HAD。鉴于CF可影响患儿胰腺外分泌功能不全,末端回肠黏性粪便积聚引起慢性DIOS,患儿腹胀、营养不良、结直肠萎瘪等临床症状可能由胰腺外分泌功能不全及肠神经发育不良共同作用所引起。目前国内报告的CF 患者肠道手术基本没有提及肠神经节细胞发育有无异常,对于CFTR基因异常是否直接影响肠神经元发育目前尚不清楚,可通过建立CFTR 基因敲除动物模型来进一步探索两者之间的相互关系。
本例患儿病史较长,临床上以对症治疗为主,基因检测最终确诊CF,我们认为CF 患者诊断应首选汗液实验,当缺乏实验条件或氯离子浓度不能直接诊断时,选择基因检测。同时儿童CF合并HAD目前报道极为罕见,以肠梗阻为主要表现,同时合并顽固性呼吸道感染及肝功能损伤的患儿,即使术后病理提示肠神经元病理改变也应该建议尽早行汗液实验及基因检测来排除有无CFTR基因突变的可能性。
猜你喜欢
医学概论(2022年3期)2022-04-24
上海护理(2021年4期)2021-12-01
天津医科大学学报(2021年4期)2021-08-21
中国民间疗法(2020年22期)2021-01-07
中国内镜杂志(2017年2期)2017-03-20
中国当代医药(2015年1期)2015-03-01
中国卫生标准管理(2015年3期)2015-01-27
中国中医药现代远程教育(2014年15期)2014-03-01
中国中医药现代远程教育(2014年14期)2014-03-01
中国中医药现代远程教育(2014年13期)2014-03-01
