
基于生物信息学方法分析影响胆管癌发生发展的差异基因
2022-12-17 06:10王玉明邵可刘一纬吴晓峰
南京医科大学学报(自然科学版) 2022年12期
王玉明,邵可,刘一纬,吴晓峰
南京医科大学第一附属医院肝胆中心,江苏 南京 210029
胆管癌(cholangiocarcinoma,CCA)起源于肝内或肝外胆管上皮细胞,根据解剖位置分为肝内胆管癌(intrahepatic cholangiocarcinoma,iCCA)、肝门部胆管癌(perihilar cholangiocarcinoma,pCCA)和远端胆管癌(distal cholangiocarcinoma,dCCA)[1-2]。在大多数CCA中,pCCA 占60%~70%,dCCA 占20%~30%,iCCA 占5%~10%。CCA 是第二常见的肝胆恶性肿瘤,占所有肝胆恶性肿瘤的1%~15%[3]。尽管CCA的平均发病率较低,但CCA的早期诊断和治疗难度较大,患者整体预后较差[4]。最近,iCCA 已成为原发性肝肿瘤相关死亡的主要原因[5]。目前全身药物治疗仅限于晚期或转移性CCA 患者,而手术治疗仅适用于早期CCA 患者,其复发风险高。中晚期CCA 患者中位生存时间少于2年,5 年生存率仅为10%[3,6]。寻找影响CCA发生和进展的基因驱动因素对于探索分子诊断和靶向治疗具有重要意义[1]。近年来,生物标志物研究在CCA的预测、治疗和预后方面取得了进展[7]。例如,在pCCA和dCCA中已鉴定出KRAS突变和PRKACB 融合基因,在iCCA中已鉴定出异柠檬酸脱氢酶(IDH)的体细胞突变[8]。此外,诱导型一氧化氮合酶(iNOS)通过炎症依赖性方式参与CCA的发生。然而,由于遗传异质性强,目前对CCA分子机制的认识尚不全面,对促进CCA启动和发展的遗传变异的理解仍然是片面的。此外,致癌的关键驱动基因仍然未知[4,9]。因此,研究CCA的发病机制并确定参与CCA发展的枢纽基因仍然是一项重大挑战。
基因综合表达数据库(gene expression omnibus,GEO)是一个公共基因表达库,包含超过94 000 个数据集和超过200 万个样本[10]。这是一个丰富的资源库,通过适当的方法和工具,可用于整合基因表达数据,用于生物标志物发现[11]、疾病分类或表型比较[12]等。癌症基因组图谱(TCGA)是一项公共资助项目,主要目的在于对30多个人类癌症的大型队列中的主要致癌基因组改变进行研究。有许多基于TCGA 的CCA肿瘤学研究[15-16],如Wang等[17]研究了lncRNA-miRNA-mRNAceRNA 网络,并确定了3 种lncRNA,即COL18A1-AS1、SLC6A1-AS1 和HULC与CCA 患者的总体存活率显著相关。为了对癌症基因组图谱进行综合分析,在TCGA 中应用了依赖于使用微阵列和下一代测序方法的高通量技术[13]。RNA 测序(RNAseq)已成为转录组(总RNA)分析和获取准确链信息的有用工具[14]。RNAseq 是一种有利于对差异表达基因相互作用和相关信号通路进行高精度系统综合研究的方法。此外,蛋白质-蛋白质相互作用网络(protein-protein interaction,PPI)可用于区分中枢基因,这些基因被定义为具有高度连接性的基因,在稳定PPI 网络结构方面发挥重要作用。
本研究旨在通过运用生物信息学方法分析GEO数据库中基因芯片数据集,发掘CCA的核心基因,为探索CCA的发病机制和潜在治疗靶点提供一定的理论依据。
1 资料和方法
1.1 资料
本研究所使用的基因数据来自基因表达数据库GEO(https://www.ncbi.nlm.nih.gov/geo/)。从数据库中检索关于人CCA 相关的数据集,经过筛选,选择了3 个基因表达谱(GSE32879、GSE45001和GSE76297),其中GSE32879 和GSE76297 分别来自Affymetrix 的GPL6244和GPL14550,GSE45001来自Agilent的GPL14550(表1)。
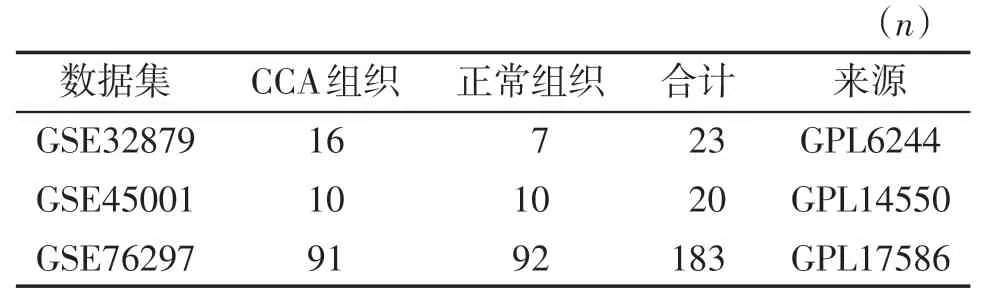
表1 CCA基因芯片数据集基本信息Table 1 Basic information of gene chip data sets for CCA
1.2 方法
1.2.1 差异表达基因(differentially expressed gene,DEG)的获取
使用在线分析工具GEO2R(https://www.ncbi.nlm.nih.gov/geo/geo2r/)分析上述3 个数据集中CCA样本和正常样本之间的DEG,以P<0.05 且|log2FC ≥1|的基因被认为是DEG。
1.2.2 差异基因的火山图和韦恩图的绘制
在线分析使用韦恩图网络工具(bioinformatics.psb.ugent.be/webtools/Venn/)对3个数据集的DEG取交集而获得共有差异基因。
1.2.3 DEG的GO和KEGG通路分析
GO 分析是功能富集研究的常用方法之一;KEGG 是一个被广泛使用的数据库,它存储了大量关于基因组、信号通路、疾病模型和药物等的数据。本研究中DEG的GO注释富集分析和KEGG通路富集分析是通过DAVID 数据库工具(https://david.ncifcrf.gov/)进行的。P<0.01和计数≥10认为具有统计学意义。
1.2.4 蛋白互作网络和枢纽基因的构建
使用检索相互作用基因(STRING)数据库(http://string-db.org/)进行PPI分析。随后,使用Cytoscape软件(www.cytoscape.org/)对PPI进行可视化,并构建枢纽基因模块。在本研究中,前10个基因被确定为枢纽基因。
1.2.5 枢纽基因的表达验证及生存分析
运用GEPIA(http://gepia.cancer-pku.cn/)对枢纽基因进行表达水平和生存分析。
1.2.6 枢纽基因的表达验证
通过RT-PCR 和Western blot 检测枢纽基因SLC2A2在CCA及正常胆管细胞的表达量(SLC2A2-F:5′-GCTGCTCAACTAATCACCATGC-3′;SLC2A2-R:5′-TGGTCCCAATTTTGAAAACCCC-3′)。
1.3 统计学方法
SPSS 20 用于数据处理和统计分析。每组3 个独立重复的数据表示为均数依标准差()。两个独立样本组间比较采用t检验,多组间比较采用方差分析(ANOVA)。生存分析采用Kaplan-Meier法并进行Log-rank检验,P<0.05 为差异有统计学意义。
2 结果
2.1 DEG的筛选
使用在线数据库对差异基因进行火山图绘制(图1A~C)。根据P<0.05 和|log2FC|≥1 的标准,从GSE32879中共鉴定出573个上调和1 167个下调基因;在基因芯片GSE45001中共鉴定出667个上调基因和1 481 个下调基因;GSE76297 芯片中共鉴定出416个上调基因和586个下调基因。随后,对3个数据集取交集绘制韦恩图(图1D)。最后,筛选出151个上调基因,50 个下调基因,选择上调基因进行后续的分析研究。

图1 差异基因火山图及韦恩图Figure 1 Differential gene volcano map and Wayne map
2.2 DEG的功能富集分析
使用DAVID对上调的差异基因进行GO功能和KEGG 通路富集分析。GO 富集分析主要包括生物学过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)3 个方面。BP 主要集中在氧化还原反应和药物反应;CC方面,主要作为线粒体、线粒体基质、胞质、外泌体等发挥作用;MF 分析提示DEG 在肽链内切酶活性和受体结合中发挥作用(图2A)。此外,KEGG通路分析结果表明差异基因主要集中在代谢、补充凝固级联和过氧化物酶体等信号通路中(图2B)。
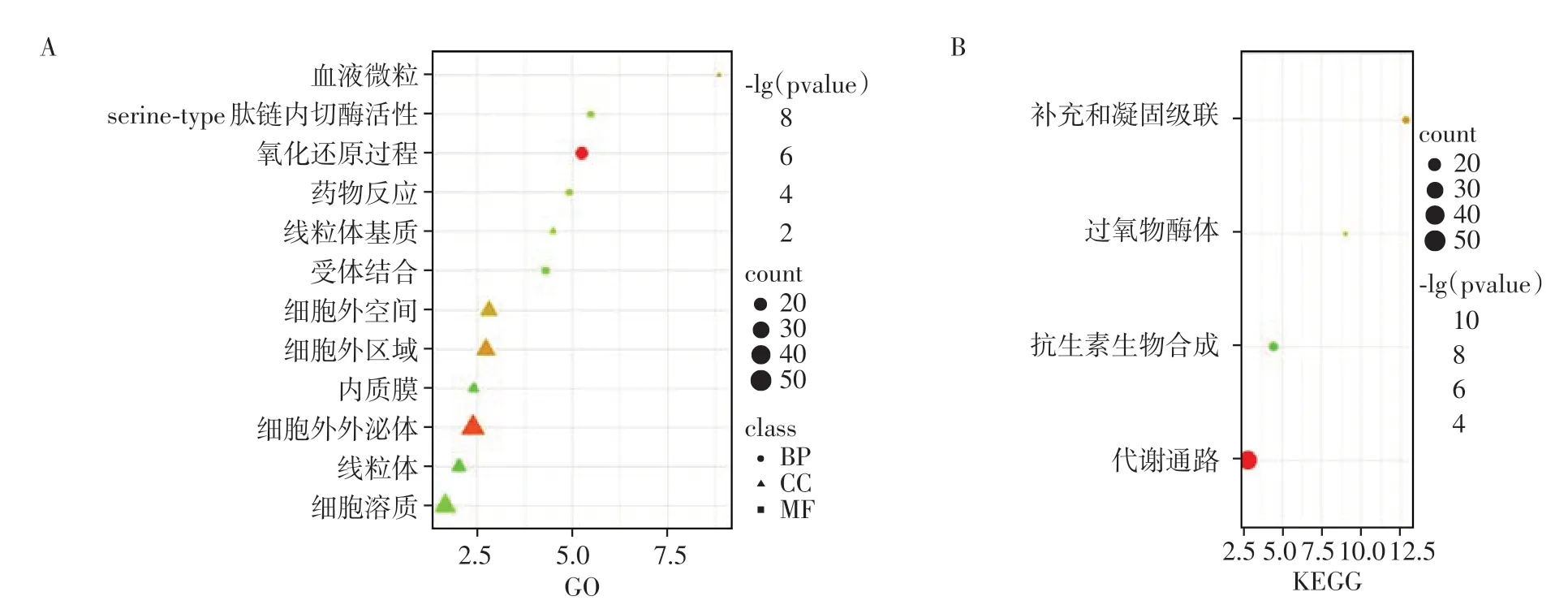
图2 CCA相关基因的富集分析Figure 2 Enrichment analysis of genes associated with CCA
2.3 PPI网络分析和枢纽基因模块构建
使用STRING工具预测DEG之间的蛋白质相互作用,将获得的PPI 数据导入Cytoscape 软件并运用CytoHubba 插件计算每个蛋白之间的连接度,筛选出连接度最高的前10名作为枢纽基因(图3)。结果表明,甲酰亚胺基转移酶环脱氨酶(FTCD)连接度最高(为35),其他枢纽基因依次是AGXT、SERPINC1、FETUB、F13B、SLC2A2、APOA1、HGD、PIPOX 和CAT,连接度分别为34、28、2、25、25、23、23、22 和2。所有这些枢纽基因在CCA中均被上调。
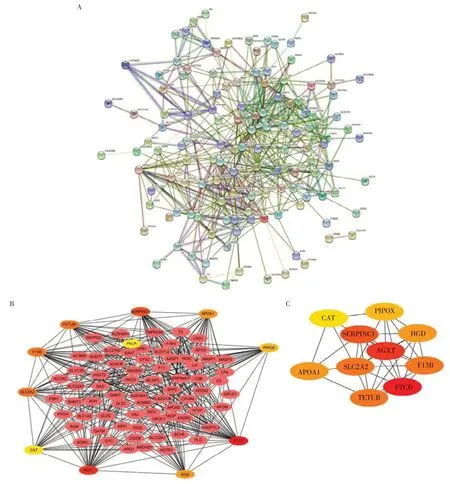
图3 差异基因蛋白互作网络分析图Figure 3 Differential gene protein interaction network analysis diagram
2.4 枢纽基因的生存分析
为了研究10个潜在枢纽基因的预后价值,使用了GEPIA 生物信息学分析平台。共有36 例CCA患者可用于总生存期的分析。结果发现,在这些枢纽基因中,有6 个枢纽基因的高表达与CCA患者不利的总体生存率有关(图4)。由于TCGA 数据库中的CCA患者仅有36例,因此进行生存统计学分析时生存数据无统计学意义,但总体趋势有差异。
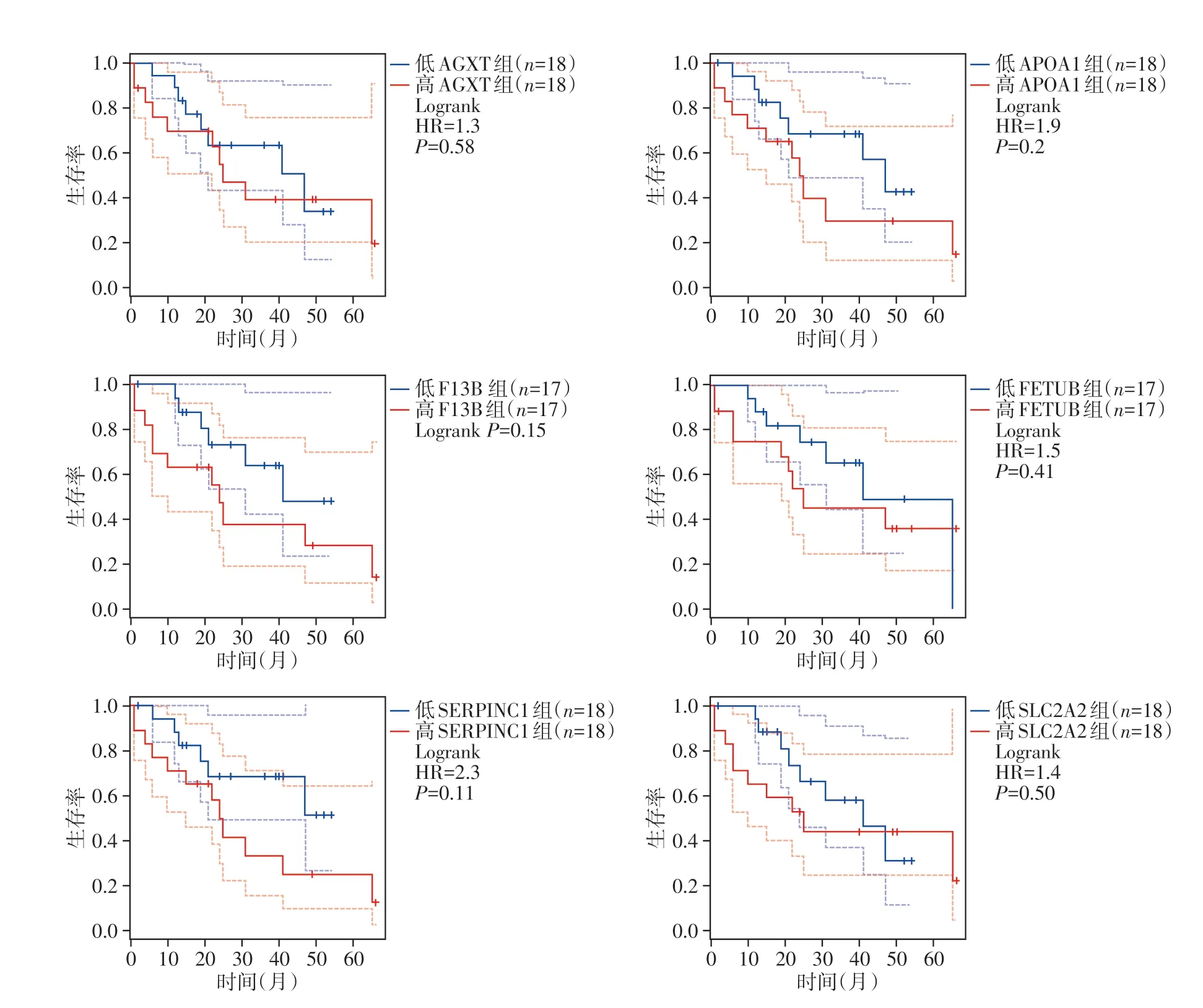
图4 6个胆管癌枢细基因与患者总体生存率分析Figure 4 Survival analysis of hub genes in cholangiocarcinoma in TCGA
2.5 枢纽基因在CCA组织表达与免疫浸润水平
肿瘤浸润淋巴细胞影响各种癌症患者的生存。因此,分析与预后相关的枢纽基因表达与6 种浸润性免疫细胞(CD8+T 细胞、CD4+T 细胞、B 细胞、树突状细胞、巨噬细胞和中性粒细胞)的关联。分析结果显示在CCA中,SLC2A2 枢纽基因的表达水平与B 细胞(r=-041,P=0.014)、CD8+T 细胞(r=-0.352,P=0.038)、巨噬细胞(r=-0.364,P=0.032)、中性粒细胞(r=-0.15,P<0.001)和树突状细胞(r=-0.45,P=0.006)相关(图5)。

图5 CCA 中HUB基因表达与免疫浸润之间关系Figure 5 Relationship between HUB genes expression and immune infiltration in CCA
2.6 SLC2A2在CCA细胞系的表达情况
为了验证枢纽基因的准确性,选取了SLC2A2枢纽基因进行验证,RT-PCR 和Western blot 显示SLC2A2 在正常胆管上皮细胞BEC 细胞系低表达,在RBE、CCT、9810、QCB-939 这4 种CCA 细胞系中均高表达(图6)。

图6 qRT-PCR 和Western blot 检测各细胞系SLC2A2 表达情况Figure 6 The expression of SLC2A2 in each cell line detected by qRT-PCR and Western blot
3 讨论
CCA是一种异质性疾病,各亚型的组织病理学特征和临床表现均不同。CCA 预后较差,5 年生存率不足百分之十[3]。手术切除是治疗CCA的主要方式之一[19],但CCA复发和内脏转移的可能性极高。目前,临床各种靶向药物及免疫治疗药物飞速发展,但是针对于CCA 的靶向药物匮乏,因此,对于CCA进行基础研究,了解CCA的发生发展,将为CCA的靶向治疗和免疫治疗提供新的方向。
整合多个数据集分析已被证明可以提高检测能力。与单个阵列分析相比,集成多个阵列被认为是提高结果可靠性的更好方法[20]。在目前研究中,3个数据集中的每一个肿瘤组都独立于正常组。
本研究中,基于公开数据库进行基因表达和PPI 分析,以确定与CCA 相关的潜在关键基因。基于来自GEO 数据库的基因表达谱数据筛选出CCA和健康人类胆管组织之间的差异基因。本文总共确定了151个上调的DEG 和50个下调的DEG。对上调基因做后续分析发现,上调基因GO 富集主要富集于CCA 中的氧化还原、药物反应、肽链内切酶活性、受体结合等;KEGG 通路富集分析表明,代谢途径、补充和凝固级联、过氧物酶体在内的通路存在明显富集。PPI 分析最终确定了6 个枢纽基因(AGXT、APOA1、F13B、FETUB、SERPINC1、SLC2A2)的高表达明显与更低的OS相关,在CCA中起着促进CCA 发生发展的作用。SERPINA1 和SERPINC1 是serpin 家族的成员,研究人员发现SERPINA1 可改善非小细胞肺癌细胞迁移、集落形成和抗凋亡[21]。而据报道,SERPINC1 的敲低可通过抑制PI3K抑制神经祖细胞增殖/Akt/mTOR 信号通路[22]。APOA1是高密度脂蛋白的主要蛋白质成分,它将多余的胆固醇从器官运送到肝脏进行排泄。APOA1 发挥抗细胞凋亡、抗炎和抗氧化活性,这些活性与肿瘤发生有关[23]。在恶性黑色素瘤的小鼠模型中,APOA1 也表现出抗肿瘤作用。然而,在本研究中,确定APOA1 在CCA样品中显著增加,表明APOA1 在CCA的发生发展中可能具有潜在的刺激作用,有待进一步研究。易化葡萄糖转运蛋白2(SLC2A2)因其对葡萄糖的低亲和力而被视为葡萄糖传感器,抑制SLC2A2 导致胰岛素分泌受损。研究表明,SLC2A2 在HepG2 细胞中的表达被SLC2A1取代,导致营养物质摄取增强和细胞增殖[24]。FetuinB(FETUB)是一种糖蛋白,是半胱氨酸蛋白酶抑制剂蛋白家族的一员,尤其是3 型半胱氨酸蛋白酶抑制剂之一[25]。半胱氨酸蛋白酶抑素家族基因位于小鼠16号染色体的近端区域和人染色体3q26-28上。虽然研究表明这些基因与凝血、肿瘤发生和血管生成有关[26],但是其具体功能和分子机制还是未知。过氧化氢酶(catalase,CAT)是一种250 kDa 的四聚体蛋白,由4个相似的亚基组成,每个亚基都包含一个血红素基团[27-28]。CAT在某些组织中高度表达,保护细胞免受过量形成的ROS的影响。CAT的异常表达或活性降低可导致细胞内ROS浓度升高,直接或间接诱发肿瘤发生[29-30]。结果表明,生物过程、细胞成分、分子功能和途径的变化可能在CCA的发病机制中起至关重要的作用。本研究结果强调了通过CCA 中的异常表达来调节这些关键生物学行为的重要性,这需要进一步的实验数据来进行验证。
免疫细胞是人体最重要的防御武器。免疫系统由各种免疫细胞组成,以防止入侵或传染性病原体,并消除受损细胞或癌细胞[31-32]。位于肿瘤微环境中的免疫细胞包括T 细胞、Tregs、B 调节细胞(Breg)、NK 细胞、DCs、MDSCs 和巨噬细胞等。肿瘤浸润性T 淋巴细胞是免疫系统中重要的效应细胞,可分为CD4+T细胞(辅助T细胞)和CD8+T细胞(细胞毒性T 细胞)。最终,通过数据库进行分析发现SLC2A2 枢纽基因与多个免疫细胞的浸润相关。为了验证枢纽基因的准确性,对SLC2A2 枢纽基因进行了RT-PCR和Western blot验证,结果显示SLC2A2枢纽基因在CCA 各细胞系的表达均高于正常胆管上皮细胞。
本研究分析了GEO和TCGA数据库支持的基于阵列和序列的CCA数据,在CCA中发现多个差异表达的基因和重要途径。基于这些基因进行了一系列分析,可能有助于发现新的CCA发生和发展的分子机制。本研究还有些许不足之处。首先,研究是建立在GEO 和TCGA 数据库上,数据来源和分析可能存在一些偏倚;其次,研究结果是通过统计学原理和方法所得,虽进行部分实验验证,但仍需进一步探究。
猜你喜欢
房地产导刊(2020年8期)2020-09-11
心电与循环(2020年1期)2020-02-27
商周刊(2019年18期)2019-10-12
当代陕西(2018年12期)2018-08-04
江苏农业科学(2017年5期)2017-04-15
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年3期)2015-06-10
中国医药导报(2015年24期)2015-02-28
创业家(2015年1期)2015-02-27
湖北农业科学(2014年3期)2014-07-21
