
非热等离子体协同锰铁双金属催化剂直接催化还原NO
2022-12-15 08:29谭潇齐随涛周一鸣石利斌程光旭伊春海杨伯伦
化工进展 2022年11期
谭潇,齐随涛,周一鸣,石利斌,程光旭,伊春海,杨伯伦
(西安交通大学化学工程与技术学院,陕西 省能源化工过程强化重点实验室,陕西 西安 710049)
化石燃料燃烧排放的氮氧化物(NO 占90%以上)是空气污染的主要来源之一,它会削弱臭氧层并导致酸雨和光化学烟雾的形成,对生态环境和人体健康造成严重危害[1-5],如何有效控制和减少氮氧化物的排放是解决大气环境质量问题的关键。在选择性催化还原(SCR)、选择性非催化还原(SNCR)和直接催化还原法等众多脱硝技术之中[6-7],采用氨气(NH3)为还原剂的氨选择性催化还原技术(NH3-SCR)是目前最高效、最成熟的脱硝技术[8-10]。然而,NH3-SCR 工艺中最常用的工业催化剂V2O5-WO3/TiO2存在工作温度窗口高且窄(300~400℃)、价格昂贵、有毒性等缺点[11-12]。同时,该工艺过程还伴随着未反应还原剂NH3的逃逸,有导致二次环境污染的风险[13-14]。
NO 直接催化还原法被认为是最理想的NO 脱除方法,它可直接将NO还原成对环境无害的N2和O2。与传统NH3-SCR方法相比,该方法工艺简单、无须引入还原剂且没有二次污染,满足石化生产过程的绿色、清洁以及可持续的发展需求。然而基于动力学分析可知,NO 直接分解反应需要克服较大的活化能(364kJ/mol)[15]。因此该过程不仅需要较高的温度条件,同时还需要合适催化剂的参与。经过长时间的探索研究,过渡金属及其氧化物等脱硝活性优良的催化剂受到了研究者们的广泛关注[16-17]。其中,锰氧化物催化剂不仅在较低温度呈现出优良的脱硝活性,还兼具经济无毒等环境友好特性[18-20],拥有极大的应用潜力。锰氧化物作为活性组分可被负载在具有特殊孔结构和较大比表面积的分子筛载体上,还可借助金属掺杂或等离子体修饰改性方法提高催化剂的稳定性及实用性。
非热等离子体(NTP)技术作为一种很有前景的温室气体去除方法,非常适用于低温催化协同反应过程。在非热等离子体场中,高速电子与气体分子在高压电场中碰撞变成高能离子、激发态离子以及自由基等高活性粒子,这些高能活性粒子能够在较低温度下进行下一步反应,从而降低气体分解反应所需温度[21]。近年来,越来越多的NTP技术被用于NO 直接分解脱除过程[22-25]。然而,该处理方法所需能耗较高,严重限制该技术的工业化和规模化应用,若能将热催化和等离子体技术相结合,则有望实现NO的低温高效直接分解脱除。
本文针对NO 低温直接分解的问题瓶颈,制备了SBA-15分子筛负载的锰铁双金属催化剂,同时采用非热等离子体的介质阻挡放电技术与催化剂协同提升NO低温直接分解的效率。探讨了放电功率和气体流量等影响因素与NO 直接分解的内在关系,研究了催化剂与等离子体协同作用下NO分解率随输出电压的变化,初步提出等离子体协同催化剂反应的机理,以期为氮氧化物的低温高效净化提供理论基础。
1 实验部分
1.1 催化剂的制备
所有化学品均采用分析纯。MnFe/SBA-15 催化剂采用浸渍法进行制备,其中,Mn和Fe的理论质量分数分别控制为10%和0.5%。将0.2g Mn(NO3)2(50%, Aladdin) 溶 液 和0.036g Fe(NO3)3·9H2O(99%,Macklin)加入到50mL 去离子水中混合,接着称取1g SBA-15(北京百灵威科学有限公司)分子筛加入上述混合溶液中,超声搅拌1h 后静置24h。然后将其在鼓风干燥箱中60℃下干燥12h,得到催化剂前体。最后,将前体在马弗炉中500℃下焙烧4h,即得MnFe/SBA-15催化剂。
1.2 催化剂的表征
XRD测试采用日本岛津公司XRD-6100射线衍射仪(Cu Kα射线,λ=1.54178Å,1Å=0.1nm),以研究催化剂的活性组分,操作电压为40kV,操作电流为30mA,扫描范围(2θ)为10°~80°,扫描速度为10°/min。
N2吸附-脱附测试采用日本麦奇克拜尔公司的BELSORP-Max全自动快速比表面及孔隙度分析仪,以研究催化剂的比表面积和孔结构参数。测试时取0.05g样品,在200℃下脱气2h,脱气完毕后,以N2为吸附质,于-196℃下进行测定。用Brunauer-Emmett-Teller(BET)方法计算样品的比表面积,用Barrett-Joyner-Halenda(BJH)方法根据N2脱附等温曲线计算孔体积、平均孔径和孔径分布。
SEM 测试在美国MAIA3LMH 型扫描电镜上进行,以观察催化剂的微观形貌。测试前需对样品进行喷金处理,以增加材料的导电性。主要测试参数为:分辨率0.7nm,放大倍率为46~1000000倍,加速电压为1~30kV。使用Aztec X-MaxN 50mm2能量色散光谱(EDS)系统进行元素映射以获得样品的成分,加速电压为16kV。
TEM 测试在日本JEM-2100 透射电镜上进行,以观测催化剂样品微观结构。主要参数为:点分辨率0.24 nm,线分辨率为0.14nm,最小束斑尺寸0.5nm,加速电压80~200kV,最高放大倍数为150万倍。
XPS 测试仪器采用美国Thermo Fisher ESCALAB Xi+型X 射线光电子能谱仪,研究催化剂的表面物种组成和化学态变化,测试条件为:真空度5×10-10mbar(1bar=105Pa),Mg Kα射线(hν=1253.6eV),C1s校准结合能为284.6eV。
1.3 催化剂的活性评价
非热等离子体协同MnFe/SBA-15 催化剂还原NO 的实验在可加热型介质阻挡放电同轴式反应器(DBD,南京苏曼等离子体科技有限公司)中进行。该反应器的阻挡介质采用内径为20mm 刚玉陶瓷管,并配备直径为12mm 的同轴高压不锈钢电极。采用石英棉将0.3g催化剂(60~80目)包覆在高压电极棒上并置于等离子体放电区域正中,与等离子体通过一段式结合。等离子体放电功率和放电电压由CTP 2000K型等离子体实验电源调节。活性测试反应气体为300ppm(1ppm=1µL/L)NO,20%(体积分数)空气(使用时),Ar为平衡气,气体总流量为200mL/min。所以反应均在25℃下进行,反应前先采用反应气吹扫整个气路30min,随后开始反应,反应后气体浓度采用Testo340烟气分析仪进行分析。NO转化率(X)计算如式(1)。
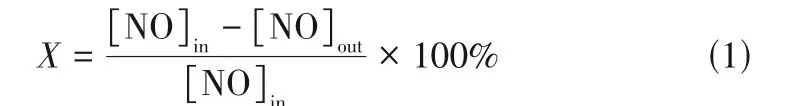
式中,[NO]in为反应器进口NO的浓度,µL/L;[NO]out为反应稳定后出口NO的浓度,µL/L。
采用德国Pfeiffer 公司的GSD320 02OmniStar 在线质谱仪分析出口气体的组成变化。在线质谱分析在等离子体反应器普通状态(0kV)下出口信号值达到稳定后开始进行,逐步提高等离子体放电电压,出口成分信号值的变化代表其相对含量的变化。实验中放电电压、放电频率和功率由一台带有1000倍衰减探针的示波器(TBS1102,鼎阳科技有限公司)进行测量,测量电流的取样电阻为50Ω。等离子体的放电功率根据式(2)计算。

式中,P为等离子体的放电功率,W;U1为放电电压,V;U2为取样电压,V;R为取样电阻,Ω。
能量密度(SIE)是研究低温等离子体过程中放电对反应物分解影响的重要参数,是等离子体处理能力最直观的体现。非热等离子体的放电能量密度(SIE)根据式(3)计算。

式中,Q为气体流速,L/s。
2 结果与讨论
2.1 非热等离子体分解NO影响因素
非热等离子体与MnFe/SBA-15 催化剂协同作用下放电功率和气体流量两大因素对NO分解的影响结果如图1 所示。图1(a)为气体流量为200mL/min时,不同等离子体放电功率下NO转化率随放电时间的变化趋势。当放电功率为2W 时,NO 基本没有分解,放电功率增大为4W 时,NO 转化率大幅增加为67.4%,说明此时已达到等离子体分解NO的能量输出功率需求。随着等离子体功率不断增大,同一时间内NO转化率逐步增大;当固定放电功率不变时,NO转化率则先随时间增加逐渐增大,最后趋于稳定。比较上述结果发现,特定放电功率下NO转化率存在极限值,提高等离子体的放电功率则会明显提升NO 转化率。当放电功率为10W时,15min 后NO 分解率即可稳定到达95%以上。在固定等离子体放电功率条件下,气体流量对NO转化率的影响结果如图1(b)所示。相同反应时间内,虽然气体流量降低有助于NO转化率提升,但气体流量的降幅变化并未显著影响NO转化率,当气体流量降低60%(从200mL/min 降低为80mL/min)时,NO 转化率提升幅度低于20%。此外,对比图1(a)和(b)中相同能量密度(3000J/L)的两条曲线发现,等离子体放电功率越大,NO 转化率越高。这表明在能量密度数值相同时,放电功率相对流量对等离子体分解NO效率影响更为显著。

图1 等离子体协同催化时能量密度两大因素对NO转化率的影响
2.2 非热等离子体协同催化剂活性评价结果
图2为不同电压下介质阻挡等离子体单独和协同MnFe/SBA-15催化剂直接分解NO的结果。由图可知,等离子体单独作用于NO分解时,放电电压升至8kV时NO转化率仍仅为3.1%;继续升至10kV时,NO 转化率提高至63.6%。相比之下,等离子体协同MnFe/SBA-15催化剂分解NO的转化率大幅提升。当电压为8kV 时,NO 转化率可达到85.3%;而当电压为12kV 时,转化率已达到97.8%,相当于等离子体单独作用时20kV 的效果(97.7%)。以上结果表明,催化剂的加入能大大提高NO在低温下的转化率,显著降低等离子体分解NO能耗。
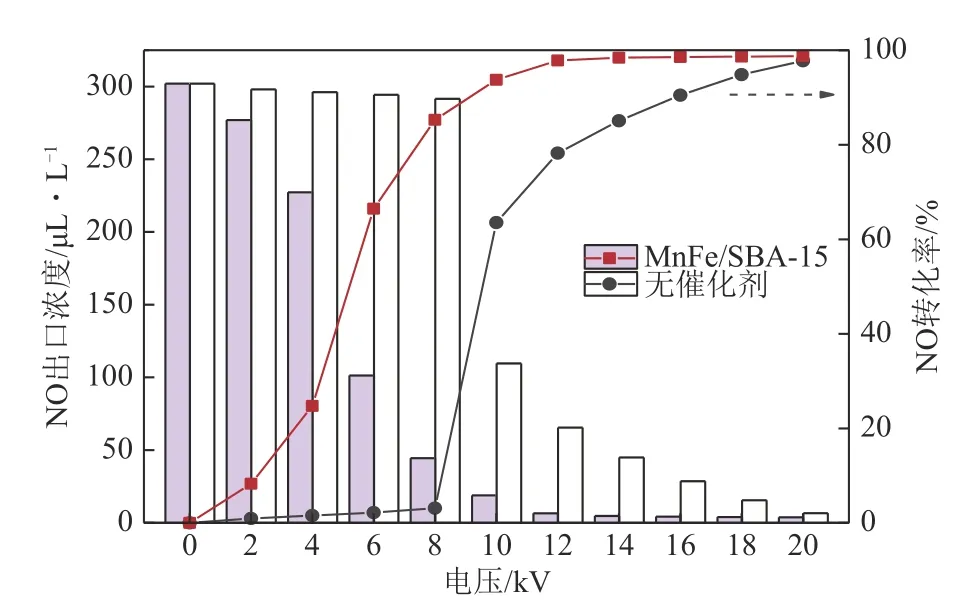
图2 不同电压下等离子体单独和协同MnFe/SBA-15催化剂对NO转化率的影响
非热等离子体协同催化剂活性评价结果表明,在0~20kV放电电压(反应电压每10min增加一次,每次增加2kV)下,等离子体协同催化剂的脱硝活性远远高于等离子体单独作用。为研究该放电条件下等离子体对催化剂理化性质的影响,将该条件下反应后的MnFe/SBA-15 催化剂与新鲜MnFe/SBA-15催化剂进行了XRD、SEM、TEM、BET、XPS 等一系列对比表征测试,结果如图3所示。
2.3 X射线衍射分析
图3(a)是MnFe/SBA-15催化剂协同非热等离子体催化反应前后的XRD 谱图。由图可知,反应前后MnFe/SBA-15 催化剂的特征衍射峰未见有明显区别,表明等离子体条件下催化剂的晶相组成未发生变化。图中2θ为28.6°、37.3°、42.8°、56.6°和72.2°等处的特征衍射峰分别对应软锰矿(β-MnO2)结构的(110)、(101)、(111)、(211)和(301)衍射面(PDF#81-2261),而2θ为32.9°和35.7°处的衍射峰归属于Mn2O3结构的(222)和(123)衍射面(PDF#76-0150)。其中,MnO2的特征衍射峰占主要部分,衍射峰强度大且较为尖锐,表明催化剂主要以MnO2晶型为主,而Mn2O3则主要以无定形态锰氧化物形式存在。此外,MnFe/SBA-15 催化剂的XRD 谱图中未检测到与Fe 相关的特征衍射峰,这可能与Fe含量较小且均匀分散有关。
2.4 N2吸附-脱附分析
MnFe/SBA-15催化剂反应前后的N2吸附-脱附等温线及相应孔径分布曲线如图3(b)所示。催化剂的N2吸附-脱附等温线为典型的Ⅳ型吸附等温线,具有明显的滞后环,表明催化剂存在介孔结构。表1列出了MnFe/SBA-15 催化剂的比表面积、孔容及孔径分析结果,可以看到MnFe/SBA-15催化剂具有较大的比表面积(446.82m2/g)和孔容(0.66cm3/g),平均孔径为6.42nm。较大的比表面积有利于NO气体分子在催化剂表面的吸附,这为NO分子提供了反应场所。在等离子体协同催化反应后,催化剂的比表面积(410.15cm2/g)、孔容(0.66cm3/g)及平均孔径(6.40nm)并未发生太大变化。这表明在较高输出电压的等离子体作用下,催化剂的多孔结构并未遭到破坏,这与扫描电镜所得结果一致。

表1 MnFe/SBA-15催化剂的BET比表面积、孔容及孔径
2.5 扫描电镜及透射电镜分析
利用扫描电子显微镜(SEM)和透射电子显微镜(TEM)对MnFe/SBA-15 催化剂反应前后的微观结构进行了表征。从图3(c)和(d)中可以看出,SBA-15 分子筛载体的结构呈并列香肠状,每段长度约为2µm,载体内部存在许多均匀并列的空隙,Mn、Fe金属形成的氧化物颗粒均匀负载在SBA-15表面及空隙间,这种立体密集分布有利于活性位点的暴露,可增加反应物与活性组分的接触概率,提高反应活性。由图3(e)催化剂反应后SEM图可以看到,催化剂结构未见太大变化,说明等离子体工作电压范围内催化剂的结构未被破坏。催化剂反应后的TEM图[图3(f)]中,MnOx以纳米颗粒分散于SBA-15载体上,可发现清晰的不同方向的β-MnO2(110)晶格条纹,但与标准β-MnO2(110)面(0.240nm)相比略大,其条纹间距在0.243nm左右,这一结果与XRD 分析一致。这可能是由于MnO2中的部分Mn4+被离子半径较大的Fe3+取代的缘故。
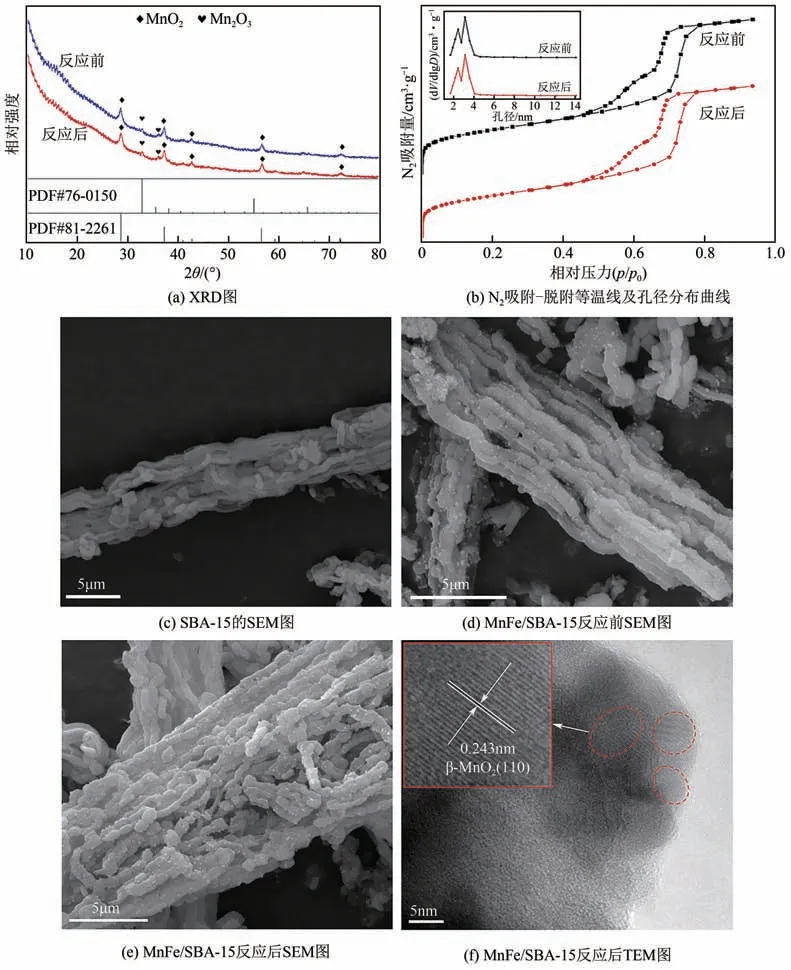
图3 MnFe/SBA-15催化剂反应前后表征结果对比图
2.6 X射线光电子能谱分析
MnFe/SBA-15催化剂的XPS分析结果如图4所示。图4(a)为Mn 2p XPS 谱图的高斯拟合分峰处理结果,其中,结合能为640.7eV、642.0eV 和644.1eV 的峰分别对应Mn2+、Mn3+和Mn4+物种[26-28]。不同价态Mn 的相对含量计算结果见表2。新鲜催化剂表面(Mn3+和Mn4+)的含量占比为73.0%,反应激活后催化剂表面的(Mn3+和Mn4+)相对含量占比仍高达69.1%,其中Mn3+占比为64.0%,高价锰(Mn3+和Mn4+)在锰物种中占比越大,越有利于锰氧化物更好实现氧化还原反应的循环[29]。丰富的三价锰离子能够与四价锰和二价锰转移电子构成氧化还原循环,提升氧化还原能力,促进NO 催化分解。图4(b)为O 1s 的XPS 谱图的高斯拟合分析结果,新鲜催化剂表面在529.7eV 和532.7eV 处的O 1s分裂峰分别对应为晶格氧(Oβ)和表面吸附氧及氧空位(Oα)物种。而反应后催化剂表面吸附氧分裂峰的结合能移动至533.0eV,晶格氧(Oβ)质量分数下降1.3%。这可能由于等离子体条件下催化剂表面因电子碰撞破坏了晶格氧稳定性,导致表面O原子脱离产生氧空位,使得表面吸附O和氧空位物种的含量相对增加。另外,Fe 原子取代也会使得MnO2中O 原子电荷不平衡,促使氧空位形成。结合表2结果可知,反应前后催化剂表面吸附氧及氧空位(Oα)质量分数从95.0%上升至96.3%。氧空位的增加改变催化剂表面电荷密度,促进电子迁移,有利于NO在催化剂表面吸附,促进NO分解。

图4 MnFe/SBA-15催化剂反应前后的Mn 2p和O 1s XPS谱图
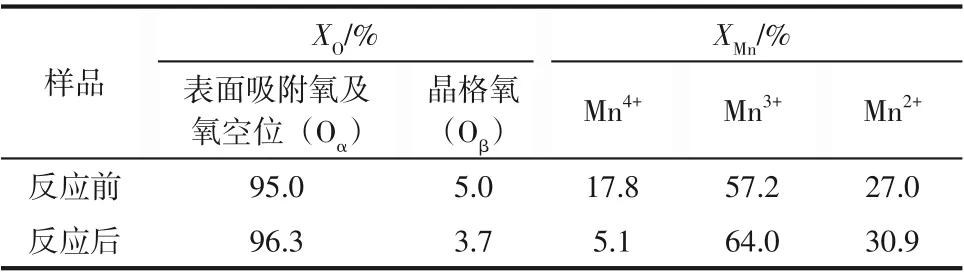
表2 MnFe/SBA-15催化剂反应前后表面不同O和Mn元素质量分数
2.7 在线质谱分析
等离子体协同MnFe/SBA-15催化剂分解NO反应过程的在线质谱分析结果如图5(a)所示。在0~8kV 间,NO 信号值逐渐降低,N2和O2的信号值相应升高,而NO2和N2O的信号值没有变化。这说明一定的放电电压才能发生NO分解反应,随着放电电压增加,NO 转化率逐渐升高。当输出电压为8kV 时,NO 转化率接近90%。反应产物在线分析结果表明,在等离子体协同催化作用下,催化剂表面吸附NO 被直接分解为N2和O2。值得注意的是,在合适的NO分解电压(4~14kV)下,尾气中N离子、O 离子的信号值变化不大,说明N—O 键断裂形成的激发态N离子、O离子吸附在催化剂上,因此可以推断此时NO分解为催化剂和等离子体的共同作用。而当电压为18kV时,尾气中N2和O2的含量急剧上升,同时伴随生成大量游离态的N 离子、O 离子,说明此时等离子体引入能量促使气相中NO 的N—O 键断裂,同时使生成的游离态N 离子、O离子无法稳定吸附在催化剂上,在气相中自发结合生成N2和O2。因此在此电压下,NO的高转化率等离子体起了主要作用。为了研究工作更贴近实际应用场景,研究了空气气氛下等离子体协同催化剂分解NO 的尾气组成,结果如图5(b)所示。可以看到,当放电电压达到10kV 时,NO 转化率开始增加。相比无空气加入时,空气气氛下等离子体协同催化分解NO的能耗明显变高。这是因为空气中O2的存在会与NO在催化剂上产生竞争吸附,使得更多的NO游离,从而增加等离子体分解NO的能耗。同时,在10~18kV 电压下,NO 含量随着电压升高依次降低,而NO2和N2O的含量逐渐升高,O2含量降低。这表明在空气气氛下NO 结合O2生成了NO2和N2O。综上,相对于Ar气氛,空气气氛下等离子体协同催化分解NO的能耗提升,同时随着反应进行,还会产生大量NO2和N2O等氧化副产物,影响反应选择性。
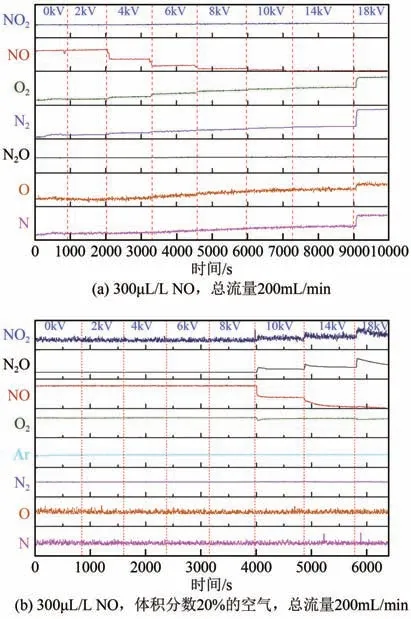
图5 等离子体协同MnFe/SBA-15催化剂分解NO的在线质谱分析结果
2.8 反应机理分析
基于催化剂活性测试结果和在线质谱分析结果,提出非热等离子体协同MnFe/SBA-15 催化剂直接催化分解NO的转化机理,如图6所示。

图6 非热等离子体协同MnFe/SBA-15催化剂直接催化还原NO机理示意图
图中反应路线1是非热等离子体单独作用分解NO,此时需要较高的输出电压才能破坏NO的N—O键,NO 分解成游离态的N 离子(N*)和O 离子(O*)后,N 离子、O 离子两两结合生成N2和O2。具体反应过程如式(4)~式(6)。

非热等离子体协同催化分解NO 的反应机理如路线2 所示。气相中的NO 分子首先吸附在催化剂表面活性位点上形成吸附态NO,Fe掺杂和等离子作用有利于在催化剂表面形成更多氧空位,为NO吸附及氧气形成提供新的活性位点。同时,不同价态Mn物种间的电子传递会削弱吸附态NO的N—O键,降低N—O 断裂所需能量。NO 在协同催化作用下解离生成N、O原子并吸附在催化剂表面,激发态N、O原子在催化剂表面两两结合生成吸附态N2和O2,最后N2和O2脱附,具体反应过程如式(7)~式(12)。

3 结论
本文通过浸渍法制备了SBA-15 分子筛负载的锰铁双金属催化剂,结合介质阻挡放电等离子体研究了等离子体协同催化低温直接分解NO 的性能。主要结论如下。
(1)活性测试结果表明,放电功率是等离子体分解NO的主要影响因素;在25℃、等离子体放电电压为12kV 时,等离子体协同MnFe/SBA-15 催化分解NO 的转化率可达97.8%,而等离子体单独作用时达到97.7%转化率所需电压为20kV。
(2)MnFe/SBA-15 催化剂的表征结果说明,等离子体协同反应前后,催化剂的晶相组成、孔道结构和物理形貌未见明显变化。XPS 分析表明Fe掺杂和等离子体作用促进了催化剂表面氧空位的形成,而催化剂上丰富的锰物种和形成的氧空位促进了NO的吸附和分解。
(3)机理分析表明,等离子体单独作用时,需要高能电场提供N—O 键断裂的能量;而当等离子体协同催化时,不同价态Mn物种间的电子传递削弱了催化剂表面吸附态NO 的N—O 键,从而降低了NO分解能耗。在降低能耗的同时,还需要关注催化剂装填与等离子体的结合方式,进一步促进二者协同作用,解决实际应用中的使用问题。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
空间科学学报(2021年6期)2021-03-09
石油石化绿色低碳(2019年6期)2019-02-13
北京航空航天大学学报(2017年7期)2017-11-24
北京航空航天大学学报(2017年7期)2017-11-24
新农业(2017年2期)2017-11-06
中国调味品(2017年2期)2017-03-20
浙江大学学报(工学版)(2016年11期)2016-06-05
Coco薇(2016年2期)2016-03-22
中国资源综合利用(2016年4期)2016-01-22
