
滑皮金柑全长转录组测序分析*
2022-12-12 12:03李智龙熊志伟高玉霞
赣南师范大学学报 2022年6期
李智龙,熊志伟,尹 辉,高玉霞,†
(赣南师范大学 a.生命科学学院; b.国家脐橙工程技术研究中心,江西 赣州 341000)
金柑(Kumquat)属于芸香科(Rutacease)柑橘亚科(Aurantioideae)金柑属(Fortunella)植物,常见品种为4种:圆金柑(Fortunellajaponica)、金桔(Fortunellamargarita)、金弹(Fortunellacrassifolia)和山金柑(Fortunellahindsii)[1].金柑为常绿灌木或小乔木,树形美观,枝条密生有短刺,叶多为卵状椭圆形,基部宽楔形.果实颜色黄亮,且含有丰富的维生素、果糖、果胶等营养成分,比其他柑橘类水果小,其整个水果包括果皮均可食用,也可以用糖浆腌制或保存,被用作传统的民间药物来治疗呼吸道炎症,为我国重要的经济水果[2-3].据文学作品记载,金柑在12世纪起源于中国,后传入日本,在中国和日本都有相当长的栽培历史,1684年由伦敦园艺协会收藏家Robert Fortune带入欧洲,不久后传入北美[4].融安金柑在中国广西已有300年种植历史,滑皮金柑为融安金柑的实生变异株.对比普通融安金柑,滑皮金柑味更甜少辣而更受消费者喜爱,是广泛的栽培品种[5].滑皮金柑已成为我国重要的经济水果,仅在融安县的种植面积达到6 272公顷,年产值达3.48亿,是当地支撑性产业[6].目前滑皮金柑的分子生物学研究几乎空白,对滑皮金柑进行组学研究有助于该品种的抗逆抗病相关研究,确保该产业的健康发展.
1 材料及方法
1.1 实验材料
2年龄滑皮金柑的叶、茎和根部样品采自赣南师范大学国家脐橙工程技术研究中心苗圃,经液氮冷冻后送至北京诺禾致源科技股份有限公司进行总RNA的提取及文库建立.
1.2 方法
1.2.1 总RNA样品检测及文库的构建
利用Trizol法对分别对金柑叶、茎和根样品进行总RNA提取[7].分别利用琼脂凝胶电泳、Nanodrop仪、Qubit荧光定量仪和Agilent 2100分析仪对总RNA的降解程度、污染情况、纯度、数量和完整性进行检测,经检测结果达到要求后混合均匀,用于文库的建立.
利用Oligo(dT)富集总RNA中含有poly(A)的mRNA,后根据SMARTer PCR cDNA Synthesis Kit使用说明将mRNA反转录为cDNA,PCR扩增富集cDNA.取其中部分cDNA利用BluePippin进行片段的筛选,富集大于4 Kb片段,并将筛选片段进行大规模PCR,以获得足够的cDNA总量.对全长cDNA进行损伤修复、末端修复、连接SMRT哑铃型接头、消化去除端末接头,构建未筛选片段和大于4 Kb片段等摩尔混合构建混合文库,结合引物、绑定DNA聚合酶,形成完整的SMRT bell文库.
1.2.2 全长转录组测序及序列分析
文库检测合格后运用PacBio Sequel平台进行测序.原始下机数据用软件包SMRT link处理获得Subreads序列,校正后获得环形一致性序列(circular consensus sequence, CCS),再根据序列是否包含5′端引物、3′端引物以及poly(A)将序列分为全长序列(Full-Length Read,FL)与非全长序列;对全长序列进行聚类,获得聚类一致序列,利用二代高质量数据对三代序列进行测序错误校正后获得高质量的一致性序列进行后续分析.
香港金柑(Fortunellahindsii)基因组已公布,是目前与滑皮金柑亲缘关系最近的物种,因此被定为全长转录组分析的参考基因组.利用GMAP软件将两者序列进行比对分析[8].根据比对结果,使用Tapis软件对一致性序列进行进一步的校正、聚类去冗余得到最终的高质量的转录本(isoform)[9].
1.2.3 全长转录组的基因结构分析
利用SUPPAV2.3软件进行可变剪接事件分析[10].使用iTAK软件进行植物转录因子预测[11].利用Tapis软件进行多聚腺苷酸化分析[9].长链非编码RNA的预测:利用PLEK和CNCI两种软件同时对转录本进行编码潜能预测,后利用CPC软件将上一步预测得到的转录本与NCBI蛋白库进行BLAST对比,进一步预测转录本[12-14].使用GMAP软件,以参数——max-intron length-ends 50000;-f 4;-z sense_force;-n 0进行融合基因的挖掘.
1.2.4 抗病基因的挖掘
结合比较基因组学和系统发生学方法系统挖掘滑皮金柑转录组中中含NB-ARC结构域抗病蛋白.具体方法是将全长基因组进行tblastn比对,筛选e-value值小于1*10-5的转录组序列进行翻译,获得含有NBS结构域蛋白.挖掘出的所有的含NBS结构域蛋白和已报道的1 601条真核及原核生物中的含NBS结构域蛋白序列一起利用hmmalign程序进行多重序列比对,得到比对序列[15].利用PhyML3.0[16]对比对文件建立系统发生关系,利用外群法确定树根.系统发生树上,与植物的含NB-ARC结构域序列形成单系群的柑橘含NB-ARC结构域序列为柑橘的NBS-LRR类抗病基因.利用HMMER(https://www.ebi.ac.uk/Tools/hmmer/)网站的phmmer功能对抗病蛋白进行结构域注释.
2 结果与分析
2.1 全长转录组测序及序列分析
通过测序,获得原始数据13.81 G.过滤原始下机数据,去除接头和长度小于50 bp的原始下机数据,得到序列,称为subreads.subreads共有10,429,790条(13.02 Gb),经测定其平均长度为1 249 bp,N50为1 556 bp.通过检测发现环形一致序列CCS共238 054个,全长非嵌合序列(Full-Length non-chimeric Read, FLNC)197 790个,占CCS的83.09%,平均长度是1 403 bp,N50为1 653 bp.使用hierarchical n*log(n)算法将同一转录本的FLNC序列进行聚类去冗余再进行校正,最终获得22 759个高质量一致序列用于后续分析.
利用GMAP软件将一致序列比对到参考基因组上,比对到基因组未注释区域的序列认为是新基因位点,比对到已知基因区域不同外显子的序列认为是新转录本.经比对分析发现总共12 040个转录本,对应6 984个基因,其中已知基因的已知转录本有5 033条,占比41.80%;已知基因的新转录本有1 418条,占比11.78%;新基因的新转录本有5 589条,占比46.42%,对应878个新基因(图1A:转录本分类图),由此可见,全长转录组发现了大量新转录本,将金柑转录本数量提升到原有的2.39倍.分析全长转录组和参考基因组转录本的长度数据,绘制长度密度图(图1B:转录本密度分布图).结果发现全长转录组利用PacBio 平台超长读长优势,与参考基因组已注释转录本相比得到长度更长的转录本序列.将参考基因已知基因的转录本和全长转录组基因的转录本数目进行比较分析,绘制成柱状图(图1C:每个基因对应转录本数目分布图).结果发现全长转录组发现更多含有2个以上转录本基因,说明全长转录组结构更完整,与参考基因组比对发现更多新转录本,补充基因注释结果.将参考基因组已知转录本和全长转录组转录本含有的外显子数量进行比较(图1D:每个转录本对应的外显子数目分布图),结果发现与参考基因组比对发现更全面的转录本外显子数量,说明全长转录组可以得到转录本更真实结构特征.

图1 全长转录组序列分析
2.2 全长转录组的基因结构分析
可变剪接,也称为选择性剪接,是某些基因的一个mRNA前体通过不同的剪接方式(选择不同的剪接位点)产生不同的mRNA剪接异构体的过程.可变剪接是调节基因表达和产生蛋白质组多样性的重要机制,是导致真核生物基因和蛋白质数量较大差异的重要原因.经鉴定,6 984个基因中共发现3 031个基因发生AS事件,包括1 099个基因发生A3(Alternative 3′ splice-site)、601个基因发生A5(Alternative 5′ splice site)、569个基因发生SE(Skipped exon)、339个基因发生RI(Retained intron)、273个基因发生AF(Alternative first exon)、123个基因发生AL(Alternative last exon)和18个基因发生MX(Mutually exclusive exon)(图2A:基因结构分析圈图,由外及内依次为:染色体序列;可变剪接位点(堆积柱状图,不同的可变剪接类型不同的颜色来表示,浅蓝色表示RI,绿色表示A3,黄色表示A5,紫色表示SE,红色表示MX,棕色表示AF,深蓝色表示AL);APA位点;新转录本分布图 ,颜色越接近于红色密度越大;新基因分布图 ,颜色越接近于红色密度越大;lncRNA密度分布;融合基因,紫色线(相同),黄色线(不同)染色体上的基因发生了融合.),说明可变剪接事件在滑皮金柑基因组中频繁发生.
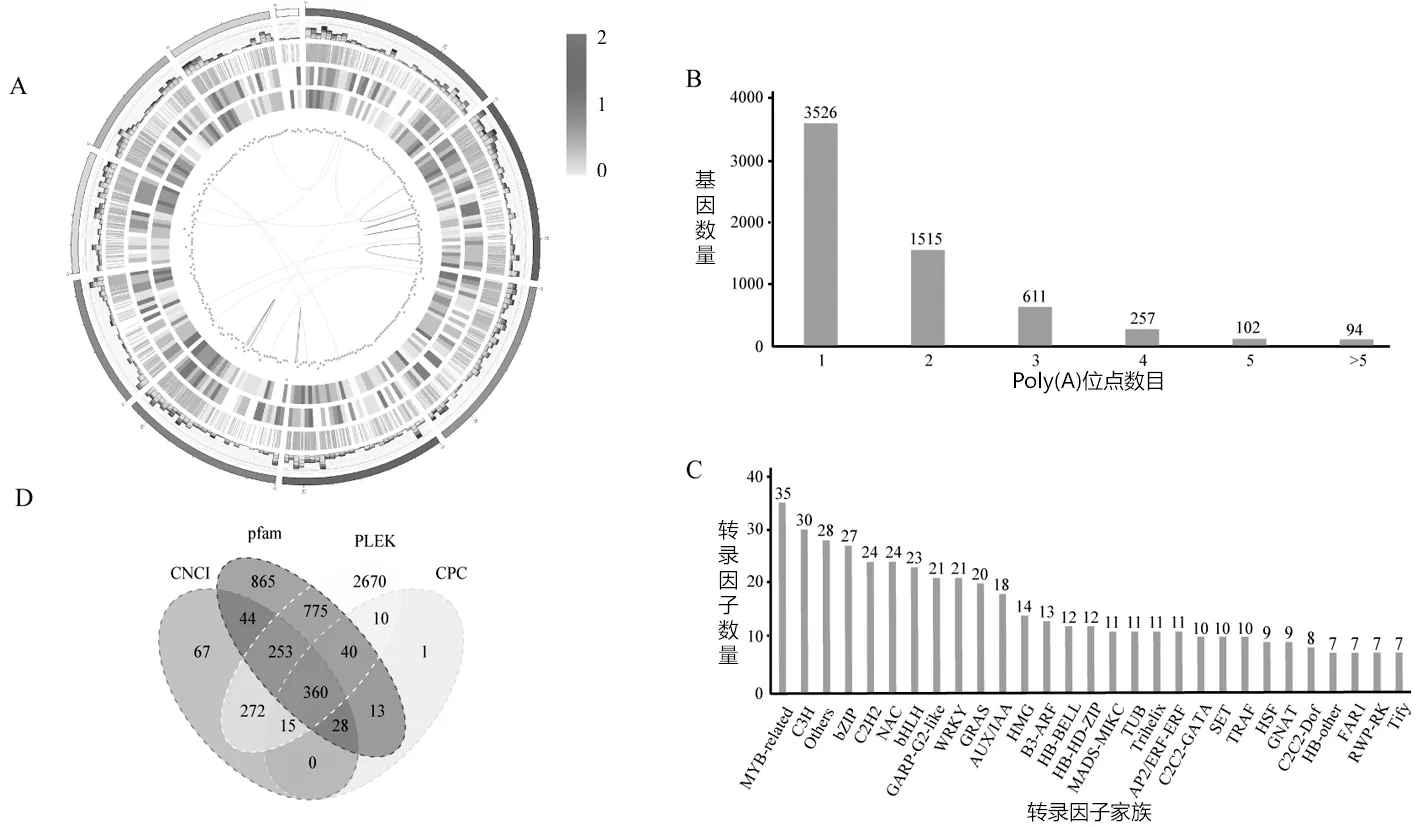
图2 全长转录组的基因结构分析图
可变多聚腺苷酸化是真核生物中一种广泛存在的基因转录后调控过程.APA在mRNA 成熟过程中对前体 mRNA 3′ 端进行加工修饰,通过调控 3′ 非翻译区 (3′ UTR) 长度而影响 mRNA 稳定性、翻译效率和定位等重要过程[17].利用Tapis软件对全长转录组进行APA检测,结果表明6 106个基因中至少含有一个poly(A)位点,其中42.25%的基因含有2个或2个以上的poly(A)位点(图2B:多聚腺苷酸化分布图),说明APA在滑皮金柑转录组中是广泛分布的.
转录因子是一群能与基因5′端上游特定序列专一性结合,从而保证目的基因以特定的强度在特定的时间和空间表达的蛋白质分子.使用iTAK软件对转录组的转录因子预测分析,结果表明:预测共得596个转录因子分布于82个转录因子家族,其中转录因子较多的家族可见图2C(图2C:转录因子分析结果图).与植物生长发育相关的常见转录因子中NAC家族有24个、bHLH家族23个、AUX/IAA家族18个、WRKY家族21个、MYB-related家族35个,这些转录因子家族的预测为之后进行金柑生长发育及次生代谢产物的合成,机体的调控提供数据基础.
长链非编码RNA是一类转录本长度超过200 nt,不编码蛋白质的RNA分子.LncRNA因具有非常重要的调控功能,且几乎参与到了各种生物学过程和通路,与各种疾病的发生发展紧密关联,从而成为过去几年和将来的研究热点和重点.通过PLEK、CNCI和CPC软件和pfam数据库的严格筛查,我们在全长转录组中共发现360个LncRNA(图2D:LncRNA预测结果维恩图).统计出mRNA的数量为11 680个,说明滑皮金柑转录组的RNA中主要是mRNA.根据LncRNA在基因组上的位置可被分为4类:基因间区的lncRNA(Intergenic lncRNA,LincRNA),主要产生于两个编码基因的中间区域;反义lncRNA (Antisense_lncRNA),主要产生于编码基因的反义链;内含子lncRNA (Sense_intronic lncRNA) 主要产生于编码基因的内含子区域;Sense_overlaping lncRNA主要位于基因内与内含子有重叠[18].对分析全长转录组中的360个lncRNA类型鉴定,发现LincRNA为122个,Antisense_lncRNA有76个,Sense_intronic lncRNA为130个和Sense_overlaping lncRNA是32个.
两个或多个基因的编码区首尾相连,置于同一套调控序列(包括启动子、增 强子、核糖体结合序列、终止子等)控制之下构成的嵌合基因称为融合基因.过去的诸多研究不断表明,基因融合与各种疾病,特别是癌症的发生发展紧密相关,甚至是一些癌症的直接诱因,所以融合基因也成为了当前组学大数据分析中的一项重要研究内容[10].依据融合基因鉴定原理,共鉴定出79个融合基因(图2A).
2.3 柑橘抗病基因挖掘
通过tblastn软件获得6条含有NB-ARC结构域蛋白,将此6序列和代表性1 601条R基因序列进行系统发生学分析,发现此6条序列均落在植物R蛋白的多样性里面,说明此6条序列均为滑皮金柑R基因,值得说明得是其中2条为新基因.利用phmmer程序对R基因进行结构域注释,发现6条R基因蛋白的结构域结构分别为RPW8-NBS、Rx_N-NBS、NBS、NBS、NBS、NBS.全长转录组中的R基因不能代表滑皮金柑中所有的R基因,而是正在发生转录的基因,所以滑皮金柑的R基因远不止6个.
3 讨论
本项目利用三代结合二代测序策略对滑皮金柑(金弹种)进行全长转录组测序,将2019年报道的香港金柑(山金柑种)基因组[19]作为参考基因组进行转录组分析,共得到12,040个转录本序列,其中41.8%为已知基因转录本,46.42%为已知基因的新转录本,11.78%新基因转录本,表明我们共挖掘出新转录本总占比为58.2%,超过香港金柑总转录本数,将金柑的总转录本数提升到原先的2.15倍.通过转录本序列分析,全长转录组利用PacBio Sequel平台超长读长优势,与参考基因组已注释转录本相比,本研究获得长度更长的转录本序列,具有更高的转录本密度;更真实转录本结构特征,以及更全面的转录本外显子数量.
本研究对全长转录组的可变剪切、多聚腺苷酸化、 转录因子、长链非编码RNA和融合基因等5个方面进行结构注释.我们发现6 984个基因中有3 031个基因发生了可变剪切事件,占比为43.4%;6 106个基因至少含有一个poly(A)位点,且其中含有2个和2个以上的poly(A)位点的基因占比为42.3%;596个转录因子分布于82个转录因子家族;360个LncRNA和75个融合基因.结果说明在滑皮金柑全长转录组中,可变剪切和多聚腺苷酸化发生频繁.我们发现转录因子预测结果多分布在金柑生长发育及次生代谢产物合成的表达部分,说明转录因子在调节金柑生长发育中发挥作用.
柑橘作为世界第一大水果,一直以来遭受严重的病害,例如柑橘黄龙病,培育柑橘抗病品种一直科学家们的研究目标.R基因在植物抵御病原入侵过程中发挥重要作用,本项目利用比较基因组学结合系统发生学方法系统挖掘了全长转录组中的R基因并对其进行结构域注释.我们共发现6个R基因,其中2条序列除含有NBS结构域外还含有rpw8和Rx_N结构域,其他四条序列只含有NBS结构域.全长转录组中的R基因不能代表滑皮金柑中所有的R基因,而是正在发生转录的基因,根据此思路可初步筛选和某一病害相关抗病基因,为滑皮金抗病机制和抗病品种的培育打下基础.
猜你喜欢
军事文摘(2022年16期)2022-08-24
湖北农业科学(2022年11期)2022-07-18
河北农业大学学报(2022年2期)2022-04-26
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年4期)2020-12-09
实用肿瘤学杂志(2020年4期)2020-12-08
中西医结合肝病杂志(2020年2期)2020-10-27
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
