
含有冠醚及偶氮苯基团的铂基菱形大环化合物的合成
2022-11-30 03:51郭大政李素婉陈茂文李一诺张榜瑞张灯青
合成化学 2022年11期
夏 炎, 郭大政, 李素婉, 李 曼, 陈茂文, 李一诺, 张榜瑞, 张灯青
(东华大学 化学化工与生物工程学院,上海 201620)
配位驱动自组装[1]是在非共价作用驱动下配体自发组装而形成有序结构的一种组装形式,在自然界中普遍存在。过去的几十年中,配位驱动自组装已经发展成为构建超分子配位化合物的有力工具,不仅涵盖了各种超分子结构的合成,而且在光化学催化[2]、化学传感器[3]、生物成像[4]和光捕获体系[5-9]等领域得到了广泛应用。其中,铂(II)超分子体系良好的光学性质和优异的自组装性能受到了研究人员的广泛关注。例如,冠醚修饰的铂(II)基金属大环在主客体化学和智能超分子材料的构建中应用较多[10]。


Scheme 1
在设计超分子大环体系的过程中,有目的地引入不同的功能基团,大环体系所表现出来的形状和性质也各不相同。冠醚是一种含有醚键的环状化合物[11],这类分子具有大小可调的空腔,环内的空腔可以捕获分子、金属离子或其它物质。受该特性影响,冠醚在主客体化学方面具有良好的应用前景[12]。偶氮苯及其衍生物通常是以反式结构存在,其在一定波长的光照射下会从反式结构转变为顺式,在可见光或者热作用下又会从顺式结构变为反式结构。由偶氮苯分子的可逆变化引起的偶氮聚合物的多种光响应特性在光学信息存储[13]、生物医学[14]和光子晶体[15-16]等诸多领域得到了广泛的应用。
本文以亚硝基苯与对氨基苯甲醇反应生成偶氮苯衍生物2, 化合物2经溴代反应得到化合物3,化合物3与3,5-二溴苯酚发生亲核取代反应得到化合物4,化合物4与4-乙炔基吡啶盐酸盐发生Sonogashira偶联反应得到化合物5。以3,6-二溴-9,10-菲醌为原料,先后经还原反应和亲核取代反应得到化合物7,化合物7与四(三乙基磷)铂反应生成有机金属铂配体8。化合物5和化合物8通过配位驱动自组装方法合成铂基菱形大环化合物1(Scheme 1),其结构经核磁、质谱和紫外可见吸收光谱表征。
1 实验部分
1.1 仪器与试剂
Persee model TU-1901型紫外可见分光光度计;Brucker Model AV-400 MHz型核磁共振仪(CDCl3、 DMSO-d6或Acetone-d6为溶剂,TMS为内标);ABSciex 4800型基质辅助激光解吸-电离飞行时间质谱仪;Micromass Quattro II triple-quadrupole型电喷雾飞行时间质谱仪。
化合物2、3[17]和6[18]参考文献方法合成;亚硝基苯、对氨基苯甲醇、四溴化碳购自上海毕得医药科技股份有限公司;三苯基膦、4-乙炔基吡啶盐酸盐、四丁基溴化铵购自国药集团;甲苯、乙醇、二氯甲烷、石油醚、乙酸乙酯、丙酮、四氢呋喃购自上海泰坦科技股份有限公司;其余所用试剂均为分析纯或者化学纯且无需特殊处理,紫外-可见光谱测试溶剂均为色谱纯。
1.2 合成
(1) 化合物2的合成
向装有磁力搅拌子的50 mL两口瓶中依次加入亚硝基苯(1.280 g, 11.90 mmol, 3 eq.)、乙醇(10 mL)、对氨基苯甲醇(491.000 mg, 4.00 mmol, 1 eq.)和乙酸(20 mL),在室温下搅拌反应4 h。之后,倒入30 mL冰水中,过滤,沉淀用硅胶柱层析(洗脱剂:二氯甲烷 ∶甲醇=50 ∶1,V∶V)纯化得橙色针状晶体化合物2659.000 mg,收率75.0%;1H NMR(400 MHz, DMSO-d6)δ: 7.88(m,J=6.9 Hz, 1.8 Hz, 4H), 7.65~7.50(m, 5H), 5.40(t,J=5.7 Hz, 1H), 4.61(d,J=5.6 Hz, 2H)。
(2) 化合物3的合成
向50 mL两口瓶中分别加入化合物2(0.314 g, 1.48 mmol, 1 eq.)、 CBr4(0.736 g, 2.22 mmol, 1.5 eq.)和磁力搅拌子,氩气保护下抽换气3次,注入新蒸THF(10 mL)。另取一干净烧杯,加入PPh3(0.582 g, 2.22 mmol, 1.5 eq.),再加入新蒸THF溶解,然后将溶解液缓慢加入到两口瓶中,室温搅拌条件下反应5 h,TLC监控反应进程。反应结束后,旋蒸除去四氢呋喃,CH2Cl2萃取(3×10 mL),合并有机相,用无水MgSO4干燥有机相,过滤除去MgSO4,旋蒸浓缩,所得粗产物用硅胶层析柱分离提纯(洗脱剂:石油醚 ∶乙酸乙酯=4 ∶1,V∶V),得到橙黄色固体化合物365.000 mg,收率90.0%。1H NMR(400 MHz, CDCl3)δ: 7.91(t,J=8.4 Hz, 4H), 7.57~7.48(m, 5H), 4.56(s, 2H)。
(3) 化合物4的合成
向装有磁力搅拌子的50 mL两口瓶中依次加入3,5-二溴苯酚(176.000 mg, 0.70 mmol, 1 eq.)、化合物3(192.000 mg, 0.70 mmol, 1 eq.)、 K2CO3(967.000 mg, 7.00 mmol, 10 eq.),氩气保护下抽换气3次,注入CH3CN(10 mL), 65 ℃油浴条件下搅拌36 h, TLC监控反应进程。反应结束后,取出反应瓶,冷却至室温,旋蒸除去溶剂,CH2Cl2萃取(3×10 mL),合并有机相,无水MgSO4干燥有机相,过滤除去MgSO4,旋蒸浓缩,所得粗产物用硅胶层析柱分离提纯(洗脱剂:石油醚 ∶二氯甲烷=2 ∶3,V∶V),得到橙黄色固体粉末284 mg,收率91.0%。1H NMR(400 MHz, CDCl3)δ: 7.95(t,J=7.4 Hz, 4H), 7.58~7.48(m, 5H), 7.29(t,J=1.5 Hz, 1H), 7.10(d,J=1.5 Hz, 2H), 5.10(s, 2H);13C NMR(101 MHz, CDCl3)δ:159.72, 152.63, 152.51, 138.63, 131.21, 129.15, 127.99, 126.97, 123.23, 122.95, 117.37, 70.01。
(4) 化合物5的合成
向装有磁力搅拌子的50 mL两口瓶中依次加入化合物4(535.400 mg, 1.20 mmol, 1 eq.)、 CuI(22.900 mg, 0.12 mmol, 0.1 eq.)、 4-乙炔基吡啶盐酸盐(502.000 mg, 3.60 mmol, 3 eq.)、 Pd(PPh3)4(138.700 mg, 0.12 mmol, 0.1 eq.),氩气保护下抽换气3次。随后注入脱气干燥的三乙胺(15 mL)和THF(15 mL),反应在惰性气氛下于80 ℃搅拌36 h,TLC监控反应进程。结束后,取出反应瓶,冷却至室温,旋蒸除去溶剂,CH2Cl2萃取(3×10 mL),合并有机相,无水MgSO4干燥有机相,过滤除去MgSO4,旋蒸浓缩,所得粗产物用硅胶层析柱分离提纯(洗脱剂:二氯甲烷 ∶丙酮=1∶1,V∶V),得到橙黄色固体粉末318.000 mg,收率54.0%。1H NMR(400 MHz, DMSO-d6)δ: 8.66(d,J=5.8 Hz, 4H), 7.95(d,J=8.3 Hz, 2H), 7.91(d,J=6.5 Hz, 2H), 7.70(d,J=8.3 Hz, 2H), 7.65~7.58(m, 3H), 7.55(d,J=5.9 Hz, 4H), 7.49(s, 1H), 7.43(s, 2H), 5.36(s, 2H);13C NMR(101 MHz, DMSO-d6)δ: 158.41, 152.61, 152.48, 149.87, 139.02, 131.21, 130.93, 129.15, 128.26, 127.90, 125.58, 123.78, 123.21, 122.93, 119.10, 92.53, 87.39, 69.84。
(5) 化合物6的合成
向100 mL两口瓶中加入六聚乙二醇(5.000 g, 47.12 mmol, 1 eq.)、 TsCl(19.000 g, 94.23 mmol, 2 eq.)、 NaOH(9.000 g, 235.60 mmol, 5 eq.)、 THF(20 mL)、 H2O(10 mL)和磁力搅拌子,冰水浴反应5 h,用TLC监控反应进程。反应结束后,旋蒸除去四氢呋喃,CH2Cl2萃取(3×10 mL),合并有机相,无水MgSO4干燥有机相,过滤除去MgSO4,旋蒸浓缩,所得粗产物用硅胶层析柱分离提纯(洗脱剂:乙酸乙酯),得到黄色油状液体化合物6.400 g,收率66.7%。1H NMR(400 MHz, CDCl3)δ: 7.73(d,J=8.3 Hz, 4H), 7.30(d,J=8.0 Hz, 4H), 4.13~4.07(m, 4H), 3.64~3.61(m, 4H), 3.58~3.50(m, 16H), 2.39(s, 6H)。
(6) 化合物7的合成
在装有磁力搅拌子的100 mL两口瓶中,依次加入3,6-二溴-9,10-菲醌(1.000 g, 2.73 mmol, 1 eq.)、 Na2S2O4(2.900 g, 16.39 mmol, 6 eq.)、 Bu4NBr(88.000 mg, 2.73 mmol, 1 eq.)、 THF(15 mL),在氩气保护下抽换气3次,搅拌10 min,然后加入氢氧化钾溶液(0.900 g, 21.75 mmol, 10 eq.),搅拌10 min,最后加入化合物6(1.600 g, 2.73 mmol, 1 eq.)。反应在惰性气氛下于80 ℃搅拌48 h, TLC监控反应进程。反应结束后,取出反应瓶,冷却至室温。旋蒸除去四氢呋喃,CH2Cl2萃取(3×10 mL),合并有机相,无水MgSO4干燥有机相,过滤除去MgSO4,旋蒸浓缩,所得粗产物用硅胶层析柱分离提纯(洗脱剂:二氯甲烷∶乙酸乙酯=10 ∶1,V∶V),得到白色固体0.870 g,收率52.0%。1H NMR(400 MHz, CDCl3)δ: 8.64(d,J=1.7 Hz, 2H), 8.16(d,J=8.8 Hz, 2H), 7.70(dd,J=8.8 Hz, 1.7 Hz, 2H), 4.44~4.39(m, 4H), 4.04~3.98(m, 4H), 3.82(dd,J=5.9 Hz, 4H), 3.76(dd,J=5.9 Hz, 4H), 3.69(d,J=4.3 Hz, 8H);13C NMR(101 MHz, CDCl3)δ:142.96, 130.55, 128.96, 128.45, 125.36, 124.40, 120.51, 72.69, 71.41, 70.81, 70.57。
(7) 化合物8的合成
向装有搅拌子的50 mL两口瓶中加入化合物7(80.000 mg, 0.13 mmol, 1 eq.),氩气保护下抽换气3次。然后加入新制Pt(PEt3)4(0.48 mmol, 3.7 eq.),甲苯(15 mL), 105 ℃条件下反应72 h,用TLC监控反应进程。反应结束后,取出反应瓶,冷却至室温,旋蒸除去溶剂,CH2Cl2萃取(3×10 mL),合并有机相,无水MgSO4干燥有机相,过滤除去MgSO4,旋蒸浓缩,所得粗产物用硅胶层析柱分离提纯(洗脱剂:二氯甲烷 ∶乙酸乙酯=3 ∶1,V∶V),得到淡黄色固体115.000 mg,收率60.0%。1H NMR(400 MHz, CDCl3)δ: 8.39(s, 1H), 7.71(d,J=8.3 Hz, 2H), 7.50(t,J=7.3 Hz, 2H), 4.38~4.32(m, 4H), 4.00~3.94(m, 5H), 3.80(dd,J=5.6 Hz, 5H), 3.73(dt,J=6.4 Hz, 5H), 3.65(s, 9H), 1.60(d,J=7.1 Hz, 28H), 1.06~0.93(m, 42H);31P{1H} NMR(121.4 MHz, CDCl3)δ(ppm): 12.35(s,1JPt-P=2770.2 Hz)。
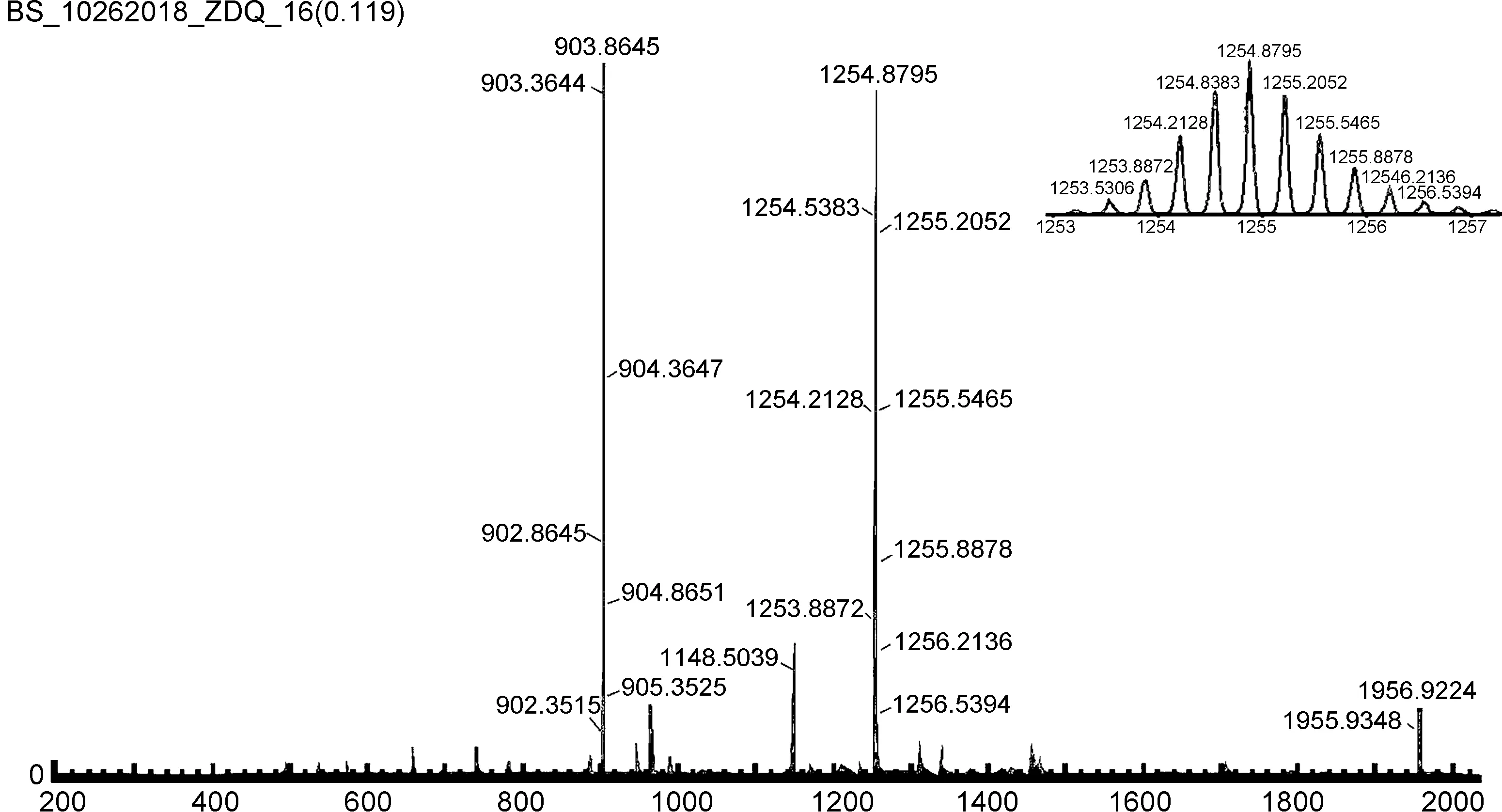
图1 化合物1的ESI-TOF Mass
(8) 铂基菱形大环化合物1的合成
向5 mL样品瓶中分别加入8(10.000 mg, 6.77×10-3mmol, 1 eq.)、5(3.300 mg, 6.77×10-3mmol, 1 eq.)和AgOTf(5.700 mg, 3.40×10-2mmol, 5 eq.),再加入3 mL新蒸的二氯甲烷,室温下避光反应12 h。停止搅拌,将反应液在2400 r/h下离心30 min,过滤,滤液用真空泵抽干得到橙黄色固体25.600 mg,收率90.0%。1H NMR(400 MHz, Acetone-d6)δ: 9.33~9.05(m, 4H), 8.84~8.67(m, 2H), 8.14~7.91(m, 8H), 7.90~7.66(m, 6H), 7.67~7.43(m, 6H), 5.48~5.37(m, 2H), 4.54(d,J=16.1 Hz, 4H), 4.09(s, 4H), 3.83(s, 4H), 3.70(d,J=14.0 Hz, 12H), 1.50(s, 18H), 1.29~1.14(m, 27H);31P{1H} NMR(121.4 MHz, Acetone-d6)δ(ppm): 13.0(s,1JPt-P=2710.2 Hz); ESI-TOF-MS: C166H224N8O16P8Pt4理论值m/z[M-4OTf]4+=903.94,实验值为903.86;理论值m/z[M-3OTf]3+=1254.94,实验值为1254.88;理论值m/z[M-2OTf]2+=1956.95,实验值为1956.92。
2 结果与讨论
2.1 合成
(1) 化合物3的合成
文献中报道的羟基溴代反应有三溴化磷低温反应法、NBS自由基溴代法、四溴化碳和三苯基磷溴代法等。本文选择了一种较温和且高效的溴代法:四溴化碳和三苯基磷溴代法。相较于三溴化磷低温反应法,该法反应条件温和。此外,相较于NBS自由基溴代法,该法反应收率更高。
(2) 化合物5的合成
参考文献合成方法[9,19],选择零价钯作催化剂,由于零价钯比较活泼易被氧化,因此在反应过程中必须严格除氧,所用溶剂需冷冻除氧后使用,反应瓶充满惰性气体保护。此外,化合物5含两个吡啶结构,在柱色谱分析时会与硅胶吸附。为了提高过柱效率,选择适量三乙胺润柱且尽量快速过柱分离。此反应在室温下基本不发生,在较低的温度下(60 ℃)收率很低,因此本文将反应温度设为80 ℃。值得注意的是,催化剂CuI的用量不宜过多,否则会增加4-乙炔基吡啶自身偶联的副产物生成。因此,催化剂用量控制在10%以下较为合适。
(3) 超分子大环化合物1的合成探究
化合物5与化合物8通过[2+2]配位驱动自组装得到超分子大环化合物1,收率90.0%。两个前驱体的化学计量比需严格控制在1 ∶1,如果称量误差较大则影响大环化合物的纯度。在组装过程中,因三氟甲磺酸银遇水潮解及见光易变质的特性,所以须在避光和干燥环境下进行组装,除此之外,加入大量乙醚洗涤可有效提高化合物1的纯度并得到干燥的固体粉末。
铂基菱形大环化合物1相较于双吡啶配体化合物5,吡啶质子氢均向低场移动,分别从7.55移动到了7.79; 8.66移动到了9.20。这归因于吡啶氮与铂配位后其电子云密度降低、屏蔽效应降低、化学位移值增加和向低场移动。31P{1H NMR}在13.01处有一单峰,说明只有一种环境的磷,且相较于化合物8其特征峰向低场移动。此外,通过ESI-TOF Mass(图1)进一步确证了大环的结构。大环化合物1的m/z[M-4OTf]4+理论值为903.94,实验值为903.86;m/z[M-3OTf]3+的理论值为1254.94,实验值为1254.88;m/z[M-2OTf]2+的理论值为1956.95,实验值为1956.92。核磁和质谱的结果表明化合物5和化合物8完成了自组装,成功合成了超分子大环化合物1。
紫外可见吸收光谱如图2所示,化合物5的最大吸收峰出现在300 nm处,这归属于偶氮苯衍生物的吸收;化合物8在259 nm处表现出较强的吸收;化合物1在波长323 nm处有最大吸收峰,归属于金属到配体的电荷跃迁。1的吸收光谱基本上是化合物5和化合物8的叠加,证明了铂基菱形大环化合物1被成功合成。

λ/nm
利用配位键驱动自组装方法,以120o给体化合物5和60o受体化合物8为原料,合成了铂基菱形大环化合物1。冠醚和偶氮苯基团的引入有利于构建特定性能的纳米组装体。该类超分子大环体系在主客体化学、光捕获、生物医学、光学器件、涂料和纺织品等领域具有潜在的应用前景。
猜你喜欢
化学工程师(2022年3期)2022-04-19
安徽化工(2022年1期)2022-02-15
上海化工(2021年2期)2021-04-23
中华养生保健(2020年3期)2020-11-16
化工学报(2020年4期)2020-05-28
化工管理(2020年1期)2020-01-16
今日农业(2019年11期)2019-08-13
中国塑料(2016年11期)2016-04-16
中国粮油学报(2016年1期)2016-02-06
科技与企业(2015年20期)2015-10-21
